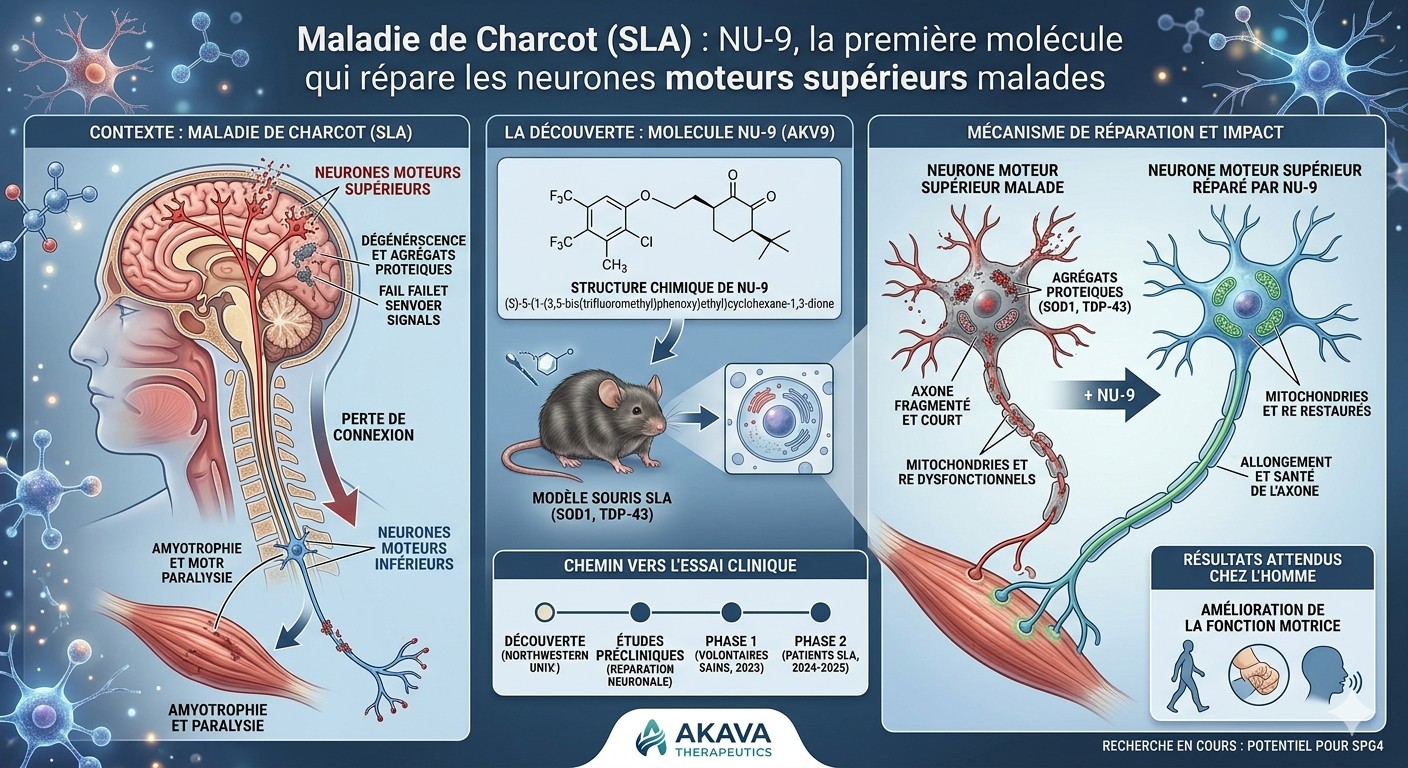
A team at Northwestern University has identified a small molecule, NU-9, capable of halting the degeneration of brain neurons that die in ALS — a first in the history of this incurable disease.
What is ALS?
ALS (amyotrophic lateral sclerosis), also known as Lou Gehrig's disease, is a neurodegenerative condition in which motor neurons progressively die. These neurons are the command chain for voluntary movement: walking, speaking, swallowing, breathing. When they disappear, muscles weaken and then become paralyzed. The disease is currently incurable, and average life expectancy after diagnosis is 3 to 5 years.
Two types of motor neurons, only one targeted until now
Motor neurons are organized in two layers. Upper motor neurons originate in the brain (motor cortex) and travel down the spinal cord. Lower motor neurons start in the spinal cord and reach the muscles. Both die in ALS — but researchers have historically focused on the lower ones, which are more accessible. The upper ones were considered "secondary." Yet in some cases they degenerate first.
What NU-9 did in this study
The researchers worked with two mouse models that develop ALS: one caused by a defective SOD1 protein, the other caused by the TDP-43 protein, which is involved in about 95% of human cases. They gave NU-9 orally every day for 60 days, starting when the mice begin showing symptoms.
The result, in numbers
5
Untreated
upper motor neurons preserved (out of ~60 in a healthy mouse)
46
With NU-9
neurons preserved, near-normal recovery
Without treatment, at 4 months, sick mice retain only 5 upper motor neurons per section observed — instead of about 60 in a healthy mouse. With NU-9, they preserve 46. The molecule doesn't just slow the disease: it halts ongoing degeneration. The mice move better too: they grip longer when flipped onto a wire grid, a test that specifically measures motor cortex neuron function.
How does NU-9 do this?
The molecule targets two problems shared by diseased neurons: their mitochondria (the cell's power plants) are broken, and their endoplasmic reticulum (the factory where proteins are made) is dilated and fragmented. NU-9 restores the structure of both. Concretely, when examined under an electron microscope, neurons from treated mice look healthy again.
⚠️ What this does NOT mean
This study was done in mice, not humans. NU-9 protects brain neurons, but not spinal cord neurons (lower motor neurons), which also die in ALS. Many molecules that work in mice fail in humans. This is therefore not an available treatment today.
And since 2021?
This foundational study was published in 2021. Since then, NU-9 has been licensed to a biotech company, Akava Therapeutics, which renamed it AKV9. The FDA cleared it for human clinical trials in August 2023. A Phase 1 study began in 2024 in healthy volunteers to assess tolerability. In 2025, preliminary Alzheimer's disease data were published in PNAS. If trials progress, a study in ALS patients could follow. It remains a long road — it often takes 10 to 12 years for a molecule to move from lab to pharmacy.
NU-9, a small molecule given orally to ALS mice, restores the integrity of mitochondria and endoplasmic reticulum in upper motor neurons and halts their degeneration in two distinct mouse models (mSOD1 and TDP-43).
Why target upper motor neurons?
Upper motor neurons (UMNs) are the corticospinal neurons of the motor cortex — Betz cells in layer V. They connect the brain to the spinal cord and orchestrate voluntary movement. Their degeneration is a diagnostic marker of ALS, primary lateral sclerosis (PLS) and hereditary spastic paraplegia (HSP). Yet at the time of this study, no molecule in clinical trials for ALS had been tested for its effect on UMNs. All current drugs (riluzole, edaravone) primarily or indirectly target lower motor neurons and associated neuroinflammation.
Two mouse models, two distinct causes
ALS does not have a single cause. The researchers tested NU-9 in two mouse models representing two independent, non-overlapping mechanisms:
prpTDP-43A315T-UeGFP: overexpresses mutant TDP-43 protein. Models TDP-43 pathology, present in approximately 95% of ALS patients.
The important nuance: these two pathologies rarely coexist. Patients with SOD1 mutations do not have TDP-43 aggregates, and vice versa. Finding a molecule effective in both situations suggests it acts on a shared cellular mechanism, downstream of the triggering proteins.
The protocol in brief
Upper motor neurons were tagged with green fluorescence (GFP) using a transgene (UCHL1-eGFP), allowing direct identification under a microscope. NU-9 was administered daily by oral gavage from postnatal day 60 (P60, symptom onset) to P120 (advanced stage), at two doses: 20 or 100 mg/kg/day. Controls received vehicle (olive oil + NMP).
Main results
Parameter (per section)
Healthy mouse
Untreated ALS model
ALS + NU-9 (100 mg/kg/d)
mSOD1 model
UMNs preserved
59 ± 3
5 ± 2
46 ± 3
Healthy mitochondria
93 %
10 %
86 %
Vacuolated apical dendrites
28 %
76 %
23 %
TDP-43 model
UMNs preserved
59 ± 4
37 ± 4
59 ± 3
Healthy mitochondria
93 %
22 %
87 %
Vacuolated apical dendrites
33 %
81 %
11 %
All differences between treated and untreated groups are statistically significant (p<0.01 to p<0.0001 depending on parameter). In the TDP-43 model, recovery is nearly complete: treated mice are indistinguishable from healthy controls on the cellular parameters measured.
Effect on motor behavior
The hanging wire test, particularly sensitive to UMN function, shows significant improvement in treated mice from P102 (mSOD1) or P120 (TDP-43), with a return to healthy mouse performance. The rotarod test, more general, shows improvement only in the TDP-43 model.
⚠️ Major limitation: no effect on lower motor neurons
In the spinal cord, lower motor neurons (LMNs) in the mSOD1 model continue to degenerate despite treatment. NU-9 does not save everything — it acts specifically on motor cortex UMNs. For human ALS, where both populations die, NU-9 alone will probably not be sufficient. Combination with other drugs (riluzole, edaravone) is explored in later publications.
Proposed mechanism
NU-9 was initially identified for its ability to reduce mutant SOD1 aggregation in cell cultures. It does reduce misfolded SOD1 levels in treated UMNs (~–28%). But its efficacy in the TDP-43 model, where there is no mutant SOD1, suggests a broader mechanism: the molecule restores shared organelles (mitochondria, ER) whose dysfunction is common to both pathologies. The precise molecular mechanism remains partially characterized — later publications (2025) suggest a lysosomal pathway.
From bench to human: what's next
This 2021 study laid the foundations. What followed is concrete: NU-9 was licensed to Akava Therapeutics (founded by Richard Silverman, the molecule's inventor, who also designed pregabalin / Lyrica). The FDA granted IND clearance in August 2023, authorizing clinical trials. Akava renamed the molecule AKV9. A Phase 1 study in healthy volunteers is evaluating its tolerability and pharmacokinetics. In 2025, results published in PNAS showed a positive effect of AKV9 in Alzheimer's disease models, suggesting a mechanism common to neurodegenerative diseases.
Genç et al. (Clin Transl Med 2021;11:e336, DOI: 10.1002/ctm2.336) show that a chiral cyclohexane-1,3-dione derivative (NU-9, 100 mg/kg/d PO, P60→P120) restores mitochondrial ultrastructure (inner membrane, cristae) and rough ER (cisternae, ribosomes), reduces mSOD1 aggregation, stabilizes apical dendrites and halts UMN degeneration in hSOD1G93A and prpTDP-43A315T mouse models, without effect on spinal LMNs.
Origin and chemistry of NU-9
NU-9 emerged from a high-throughput screen of over 50,000 compounds (Silverman Lab, Northwestern) in a dual SOD1G93A-PC12 cellular assay: protection against cytotoxicity and inhibition of aggregation. Initial hits were filtered by computational approaches (substructure searching, ligand-based clustering), then optimized through several rounds of medicinal chemistry. Hit compound 1, from the cyclohexane-1,3-dione family, led to compound 2 (good ADME but poor neuronal penetration), then to NU-9, a chiral derivative with a tert-butyl group and a bis-trifluoromethyl-phenyl group.
ADME / pharmacokinetic profile
Parameter
Value
EC50 (aggregation/toxicity)
300 nM
Aqueous solubility
≥100 µM
Microsome stability (t½)
74 min (human) / 52 min (mouse)
Plasma protein binding
90 %
PAMPA-BBB
8,11 × 10⁻⁶ cm/s (CNS+)
Brain penetration
8,3 µM
Oral bioavailability
94 %
t½ in vivo (mouse)
2,73 h
hERG / CYP inhibition (5)
None (up to 30 µM) / <10% at 3 µM
NOAEL (mouse, PO, 7 d)
100 mg/kg
Lifespan extension (mSOD1)
+13% at 20 mg/kg
Methodology: the UCHL1-eGFP reporter line
Major methodological innovation: the UCHL1-eGFP line (Ozdinler Lab, JAX 022476) selectively labels UMNs with stable GFP. Crossed with hSOD1G93A or prpTDP-43A315T, it allows in vivo identification of diseased UMNs, indispensable for quantifying a compound's cellular effects. This is the first preclinical platform enabling assessment of UMN response to treatment.
Electron microscopy: what happens inside the cell
Ultrastructural analysis by TEM (FEI Tecnai Spirit G2) on 70-nm motor cortex sections reveals the characteristic anomalies of diseased UMNs: mitochondria with disintegrated inner membrane, loss of cristae, swelling of rough ER cisternae, ribosome fragmentation. These anomalies are conserved between mouse models and human Betz cells (post-mortem samples from 9 ALS patients and 4 controls, Northwestern Brain Bank), validating translational relevance.
Mean cisterna length: 0.50 µm (mSOD1) → 1.13 µm; 0.53 µm (TDP-43) → 1.37 µm; vs WT 1.56 µm
Differential effect: UMN vs LMN
The absence of effect on LMNs in the mSOD1 model is expected and informative: at both tested doses, the number of vulnerable ChAT⁺ or NeuN⁺/ChAT⁺ LMNs does not differ from vehicle (~10–14/section vs ~46/section in WT). This specificity argues for a UMN-specific cellular target — possibly related to their morphological peculiarities (very long axons, dense cortical integration, sustained energy demand) or differential transcriptome. In the TDP-43 model, there is no significant LMN loss at P120, which prevents assessment in this population.
Critical analysis and limitations
Preclinical mouse study: extrapolation to humans remains hypothetical. The failure rate moving from ALS mouse models to clinical trials is historically high (riluzole being the exception that proves the rule).
Modest TDP-43 sample sizes: n=4 treated mice vs n=3 controls. Robust for ultrastructural effects, but limited for mortality or subtler effects.
Partial mouse models: TDP-43 pathology induced by overexpression of a mutant transgene (A315T) does not exactly reproduce the progressive cytoplasmic mislocalization observed in most patients (aggregated wild-type TDP-43).
Imprecise molecular mechanism: at the time of this publication, NU-9's direct target had not been identified. Later publications (PNAS 2025) suggest a lysosome-dependent mechanism of protein aggregation inhibition.
Not all behavioral analyses were blinded: experimenters were aware of condition (treated/untreated) during rotarod and hanging wire tests. Cellular analyses were performed blinded.
Negative rotarod effect in mSOD1 model: contrasts with positive hanging wire effect. The differential sensitivity of these tests to UMN function probably explains this result, but it limits the generalization of behavioral benefits.
Trajectory 2021–2026
Feb. 2021: publication of the founding study (Genç et al., Clin Transl Med).
March 2022: Genç et al. (Sci Rep) — NU-9 outperforms riluzole and edaravone alone, and enhances their efficacy in combination on UMN axonal regeneration.
June 2022: Akava Therapeutics licenses NU-9 (Northwestern); renamed AKV9.
July 2022: Orphan Drug Designation obtained from the FDA (section 526 FD&C).
August 2023: FDA IND clearance for AKV9 in ALS. Phase 1 in healthy volunteers authorized (single + multiple ascending doses).
Dec. 2024: $7.3M grant from the National Institute on Aging (NIH) to extend AKV9 to neurodegenerative diseases.
May 2025: PNAS publication on AKV9 and Alzheimer's disease (PMID 40030015) — 3 of 4 treated mice retained normal memory in a proof-of-concept study.
⚠️ Conflicts of interest
At the time of publication (2021), the authors declared no conflicts of interest. Since 2022, R.B. Silverman has been the founder of Akava Therapeutics, which commercializes AKV9; Northwestern University holds financial interests (royalties, equity). P.H. Özdinler is Akava's SAB Chair. These affiliations should be considered when interpreting subsequent publications from the group.
Implications and outlook
This study introduces two important concepts for developing therapies for motor neuron diseases: (1) UMN health should be a distinct preclinical endpoint, which had never been done before for compounds in clinical trials; (2) mitochondrial and ER defects converge downstream of heterogeneous causes (mSOD1, TDP-43, possibly C9orf72 and others), constituting a shared pharmacological target. The potential reach extends beyond ALS — PLS, HSP, ALS/FTLD, and now Alzheimer's. It remains to be seen whether AKV9's Phase 1, and later phases, will confirm an efficacy signal in humans.
Genç B, Gautam M, Gözütok Ö, et al. Improving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology. Clin Transl Med. 2021;11(2):e336. doi:10.1002/ctm2.336. PMID: 33634973.
Frequently asked questions
When will NU-9 (AKV9) be available for ALS patients?
Not for several years, and only if trials succeed. The FDA cleared Phase 1 in healthy volunteers in August 2023, evaluating tolerability and pharmacokinetics. A Phase 2 in ALS patients could follow, then a larger Phase 3 before any commercialization. The usual delay between first human trials and market authorization is 10 to 12 years, according to Richard Silverman, the molecule's inventor. A realistic availability would fall around 2030–2035 in the most favorable scenario.
Can NU-9 cure ALS?
The study does not suggest a cure, even in mice. NU-9 halts the degeneration of upper motor neurons (cerebral cortex), but has no demonstrated effect on lower motor neurons (spinal cord), which also die in ALS. An effective therapy would likely need to combine NU-9 with other drugs targeting lower motor neurons, such as riluzole or edaravone. Later publications by the same group (Sci Rep, 2022) explored this combination approach.
Are NU-9 and AKV9 the same molecule?
Yes. NU-9 is the academic name given by Northwestern University labs. In June 2022, the molecule was licensed to Akava Therapeutics, a biotech company founded by Richard Silverman himself, which renamed it AKV9 for clinical development. It is exactly the same chemical compound.
Why was this study only done in mice?
It is the mandatory preclinical step before any human trial. The mouse models used (hSOD1G93A and prpTDP-43A315T) were chosen because they faithfully reproduce the cellular alterations observed in the upper motor neurons of ALS patients — verified on human post-mortem samples in the same study. The move to humans requires additional safety studies (toxicology, large-animal pharmacokinetics), which Akava Therapeutics undertook before FDA IND clearance in 2023.
How does this study differ from previous ALS clinical trials?
This is the first study that explicitly evaluates upper motor neuron health as a preclinical endpoint. ALS drug candidates have historically used mouse lifespan extension or lower motor neuron survival as the main indicator. Yet many of these molecules failed in humans. Genç et al. propose a paradigm shift: target the neurons that actually degenerate (and early) in patients, and use cellular indicators (mitochondria, ER, dendrites) rather than behavioral or survival ones.
If NU-9 does not save lower motor neurons, what is the point?
First, to validate the hypothesis that it is possible to halt the degeneration of one type of motor neuron in ALS — something that had never been demonstrated. Second, because upper motor neurons play a crucial role in voluntary motor control, and are implicated in several other diseases (primary lateral sclerosis, hereditary spastic paraplegia, ALS/FTLD). Finally, in a combined therapeutic strategy: NU-9 would preserve UMNs while other drugs target LMNs. Later data (2022) even show that NU-9 enhances the efficacy of riluzole and edaravone on axonal regeneration.
Could NU-9 be effective in other neurodegenerative diseases?
Possibly. The scientific logic is sound: mitochondrial dysfunction and protein aggregation are mechanisms shared by many neurodegenerative diseases, including Alzheimer's, Parkinson's, frontotemporal degeneration (FTLD) and Huntington's. In May 2025, a publication in PNAS showed that AKV9 improves memory in a mouse model of Alzheimer's disease (3 of 4 treated mice regained normal memory). Akava Therapeutics also obtained $7.3M in funding from the National Institute on Aging in 2024 to expand research beyond ALS.
🍪 This site uses Google Analytics and the Facebook Pixel to measure audience. These tools are enabled only with your consent. Privacy policy →