When KJ Muldoon was born in 2024, he had an ultra-rare genetic disorder that kills half of affected babies in their first weeks. In 2025, an American team did something unprecedented: in six months, they designed a CRISPR treatment custom-made for this one child. Today, KJ is doing well — and medicine just crossed a threshold.
One baby, one drug, six months
When he was born in August 2024, KJ Muldoon was not expected to live long. He was born with a genetic disease affecting roughly one person in 1.3 million worldwide: CPS1 deficiency. A single misspelled letter in his DNA prevents his liver from eliminating ammonia — a byproduct of protein digestion.
Without treatment, ammonia accumulates, the brain gets damaged, and half of infants with the severe form die in the first weeks. Conventional options are poor: a hyper-restrictive protein diet buys a few months of survival; a liver transplant is only possible after several months of life.
At the Children's Hospital of Philadelphia, a team did something unprecedented. In six months, they designed, manufactured, and administered a drug custom-built for him alone. The drug is CRISPR.
What is CRISPR?
Think of your DNA as an enormous cookbook — three billion letters telling your body how to build itself. When a single letter is wrong — like a typo in the recipe — the whole dish can fail.
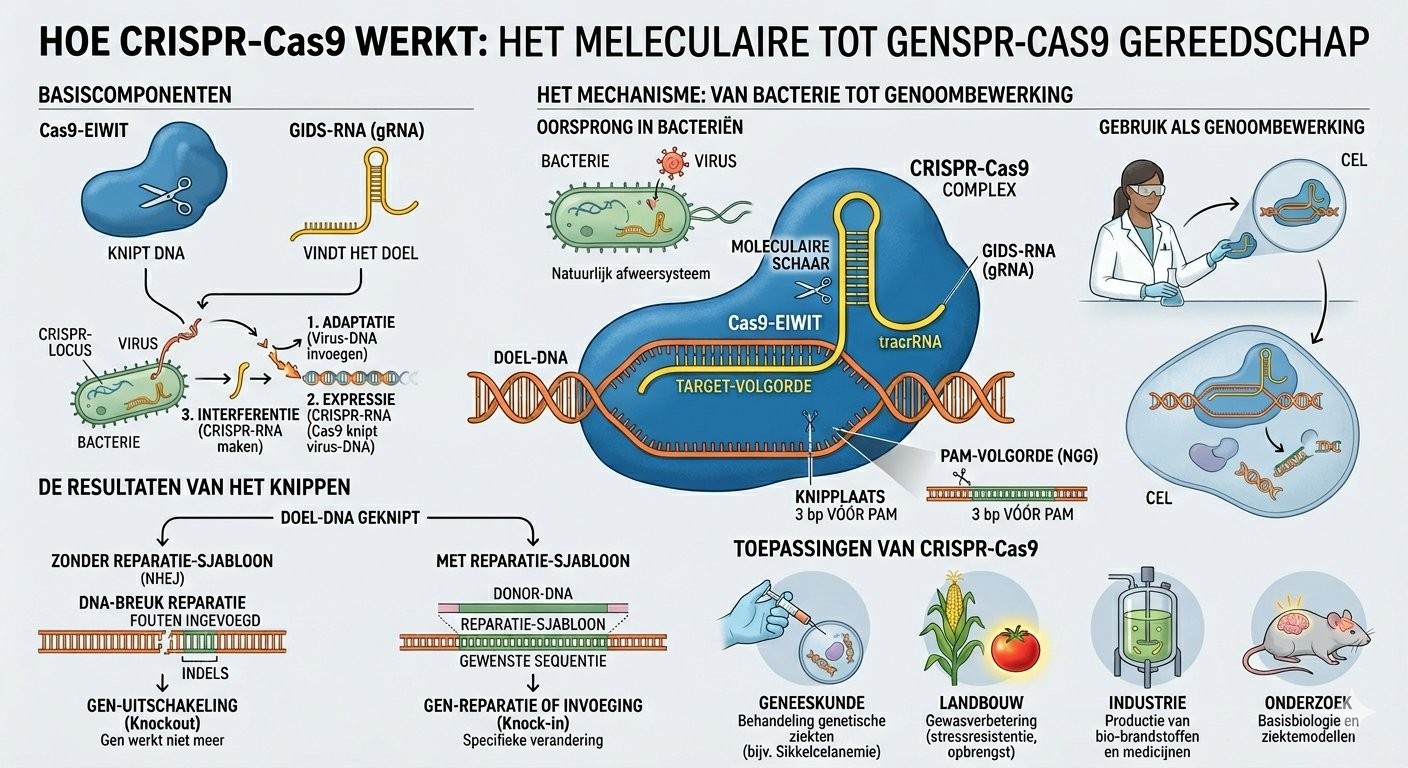
CRISPR is a pair of molecular scissors with a built-in GPS. The GPS is a small strand of RNA (DNA's chemical cousin) that can read the recipe and find exactly the sentence to correct. Once there, a protein called Cas9 cuts. The cell then handles the repair — and if guided properly, it can repair with the correct letter.
This technology has existed since 2012. It earned Emmanuelle Charpentier and Jennifer Doudna the 2020 Nobel Prize in Chemistry. What changed in 2025 with baby KJ's case is that it was used for the first time to treat a single patient unique in the world.
Why this is a historic turning point
Until now, drugs were designed for millions of people. Developing a new treatment takes ten years and costs around a billion dollars. For ultra-rare diseases — those affecting only a handful of individuals worldwide — no pharmaceutical company has an economic reason to develop anything.
The KJ case proves we can go much faster. And that the future of medicine, perhaps, is no longer a drug for everyone, but a drug for each individual.
🔍 Key takeaways
→KJ Muldoon is the first human treated with a CRISPR therapy designed specifically for him
→His disease (CPS1 deficiency) affects 1 person in 1.3 million — no lab would develop a specific drug
→The treatment was designed, manufactured, and administered in 6 months — versus 10 years typically
→KJ was discharged from hospital in June 2025 and is growing normally today
✦ To conclude
The future of medicine may be written one patient at a time.
The baby went home on June 3, 2025, after 307 days in hospital. He eats more protein than before. He needs fewer medications. He's growing. What the KJ case tells us isn't just a beautiful medical story — it's proof that genetics has crossed a threshold. Until now, rare diseases were orphans: too few patients to interest the labs. With this demonstration, the question is no longer whether we can tailor treatment, but how long and how much money it will take to make it accessible to other children.
KJ Muldoon, born in 2024 with an ultra-rare CPS1 deficiency (a urea-cycle enzyme), became in 2025 the first patient treated with a fully personalized CRISPR therapy. The team led by Musunuru and Ahrens-Nicklas in Philadelphia used base editing — a more precise evolution of classical CRISPR-Cas9 — to correct KJ's specific mutation. From design to infusion: 6 months. Published in the NEJM in May 2025.
CRISPR-Cas9: from bacterial defense to clinical medicine
CRISPR was not invented. It was borrowed. For millions of years, bacteria have used it to defend against viruses. When a virus attacks, some bacteria store a fragment of its DNA in a region of their genome called CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats). If the virus returns, a protein called Cas9, guided by an RNA copy of that fragment, finds and cuts it in two.
In 2012, Emmanuelle Charpentier and Jennifer Doudna published a paper in Science that changed everything: they demonstrated the system could be reprogrammed by replacing the guide RNA with any sequence. From then on, CRISPR-Cas9 was no longer a bacterial defense mechanism — it was a universal genome-editing tool, applicable to any living cell.
December 2023: the first CRISPR drug approved
Thirteen years after the foundational paper, the US FDA and the UK's UKMHRA approved Casgevy (exa-cel), developed by Vertex Pharmaceuticals and CRISPR Therapeutics. This treatment addresses sickle cell disease and beta-thalassemia.
The principle: the patient's stem cells are harvested, edited outside the body (ex vivo) to reactivate a gene inactive since childhood (the fetal hemoglobin gene), then reinfused. The result in treated patients: the painful crises characteristic of sickle cell disease disappear.
But Casgevy remains a "mass" treatment: the same protocol for every patient. The KJ case introduces something different.
Baby KJ: N-of-1 medicine
KJ Muldoon was born in August 2024. A few days after birth, his blood ammonia level exceeded 1,000 µmol/L — the normal range is 9 to 33. The diagnosis: severe deficiency of carbamoyl phosphate synthetase 1 (CPS1), a liver enzyme of the urea cycle. Without it, ammonia cannot be converted to urea and excreted. It accumulates. It attacks the brain.
Kiran Musunuru (University of Pennsylvania) and Rebecca Ahrens-Nicklas (Children's Hospital of Philadelphia) decided to try something else. They identified the two specific mutations in KJ's CPS1 gene, then designed a therapy tailored to those mutations. The whole thing took six months from design to infusion.
The key: they did not use classical CRISPR-Cas9. They used a more recent and more precise evolution: base editing.
Base editing: CRISPR at higher resolution
Classical CRISPR creates double-strand breaks in DNA. It's effective but risky: the cell may repair incorrectly, sometimes creating major chromosomal rearrangements.
Base editing does not cut. It chemically converts one DNA letter into another — for example an A into a G — directly in place, without a break. For the KJ case, the team designed an edit that corrects one of the two mutations responsible for his deficiency, just enough to restore a minimal level of functional enzyme.
The treatment was delivered to the liver via lipid nanoparticles (the same technology as mRNA COVID vaccines). Three doses, between February and April 2025.
Today: what we know, what we don't
What we know: KJ was discharged 307 days after admission. He can eat more protein than before. He needs fewer ammonia-scavenging medications. He is growing normally.
What we don't know: how long the effect will last, whether the edit will persist as his liver cells renew, and whether off-target effects will emerge in the years ahead.
🔍 Key points
→CRISPR-Cas9 = reprogrammed bacterial immune system as a universal editing tool (Jinek et al., Science 2012)
→Casgevy (December 2023): first approved CRISPR drug, for sickle cell disease and β-thalassemia — ex vivo approach, same protocol for all
→KJ case (February 2025): first "N-of-1" CRISPR therapy — in vivo base editing via lipid nanoparticles, targeting a unique mutation
→Historic timeline compression: 6 months from design to infusion, vs. ~10 years for a classical drug
✦ To conclude
Prototype of a model, not an isolated exception.
According to the Innovative Genomics Institute, roughly 250 clinical trials worldwide are currently using genome-editing tools, more than 150 of them active. Most are still testing uniform protocols for patient groups. But two US programs launched in 2025 (ARPA-H's THRIVE and GIVE) aim explicitly to make the "N-of-1" approach replicable: to build the regulatory and industrial infrastructure needed to compress 10 years of development into 6 months for any child with an ultra-rare mutation. If it works, it's a paradigm shift. If it doesn't, KJ will remain a magnificent but isolated exception. The next three years will decide.
Musunuru, Ahrens-Nicklas et al. (NEJM, 2025) report the first clinical case of a personalized N-of-1 genome-editing therapy in an infant with severe carbamoyl phosphate synthetase 1 (CPS1) deficiency. The team designed an adenine base editor (ABE) targeting the patient's specific truncating variants, delivered via lipid nanoparticles in three systemic doses (February–April 2025). The pipeline — from mutational identification to infusion — was completed in ~6 months. The patient tolerates more protein intake, requires fewer ammonia scavengers, and was discharged after 307 days of hospitalization.
Molecular architecture of the KJ treatment
The therapy administered to KJ is not conventional CRISPR-Cas9. It is an adenine base editor (ABE), a CRISPR-derived architecture that differs fundamentally in its mechanism of action.
An ABE is made of three fused modules:
A Cas9 "nickase" (nCas9) in which one of the two nuclease activities (HNH) has been inactivated by point mutation. It nicks only one DNA strand instead of two — avoiding the double-strand break.
An evolved adenine deaminase (ABE8e) that chemically converts an adenine (A) into inosine (I, read as guanine G by the cellular machinery).
A 20-nucleotide guide RNA complementary to the target sequence, adjacent to a PAM (NGG motif for Streptococcus pyogenes Cas9).
Concretely: the target is located by RNA-DNA pairing, one strand is nicked, and the deaminase chemically modifies a specific A within an editing window of roughly 4–8 nucleotides. After replication, the opposite strand is copied from the modified strand, fixing the A→G mutation permanently.
In KJ's case, the Musunuru team specifically targeted the truncating mutations in the CPS1 gene and designed a guide RNA enabling an A→G conversion restoring correct reading of the relevant exon. The NEJM publication (DOI: 10.1056/NEJMoa2504747) details the design and rapid preclinical testing in humanized mice before patient administration.
Why base editing changes the game
The critical difference between classical CRISPR-Cas9 and base editing lies in the safety profile. Large deletions and chromosomal rearrangements occur at a frequency roughly 20-fold lower with base editors than with classical Cas9 nucleases.
This matters because the major risks of conventional CRISPR-Cas9 are not just small off-target insertions-deletions (indels), but also broader structural variations: megabase-scale deletions, chromosomal translocations, and complex rearrangements at the editing site itself.
For a 7-month-old infant, the risk/benefit equation clearly favored the technically more conservative approach, even if it targeted a "minor" correction (partial activity restoration rather than complete correction).
Delivery: the limiting constraint
KJ's therapy was delivered via lipid nanoparticles (LNPs) — the same platform as SARS-CoV-2 mRNA vaccines. LNPs encapsulate the mRNA coding the base editor and the guide RNA, and accumulate preferentially in the liver after IV administration, which is ideal when the target (here, CPS1-expressing hepatocytes) is hepatic.
But this is also a structural limit of the model. Liver-tropic LNPs work well for the liver — hence the proliferation of trials in hepatic diseases (hATTR with Intellia's NTLA-2001, AATD-1, GSD1a with Beam Therapeutics). For other tissues (brain, muscle, retina), other vectors are needed: primarily AAV, with challenges of immunogenicity and cargo capacity (AAVs max out around 4.7 kb, tight for a full base editor).
The global clinical landscape in 2026
According to the Innovative Genomics Institute (2026 update), roughly 250 clinical trials worldwide use genome editing (CRISPR-Cas, base editing, prime editing, ZFN, TALEN), with more than 150 active. Key therapeutic areas:
The KJ case is not an isolated outlier. It fits into a broader clinical paradigm shift: from "platform" therapies (Casgevy) to "bespoke" therapies (N-of-1). The US ARPA-H launched two programs in 2025 (THRIVE and GIVE) specifically to build the regulatory and manufacturing infrastructure needed to replicate the KJ approach for other patients.
🔬 Limitations & perspectives
→Edit durability: hepatocytes renew. If editing only reached a fraction of hepatic stem cells, the therapeutic effect could erode. Long-term follow-up is crucial.
→Mosaicism: not all hepatocytes are edited. The exact editing ratio has not yet been published in detail — partial correction sufficed to restore viable metabolic function.
→sgRNA-independent off-targets: ABE8e base editors show a potentially problematic RNA editing profile. Pediatric follow-up must include transcriptomic monitoring.
→Economic scalability: treatment cost in the millions USD, covered by CHOP and philanthropic partners. No reimbursement model exists for N-of-1 therapies — the real bottleneck.
→Regulatory framework: the FDA authorized the treatment under an exceptional research protocol. Creating durable pathways for N-of-1 therapies is the 2026–2028 challenge.
✦ To conclude
The real leap is not technological — it's organizational.
CRISPR is not "the medicine of the future." It's the medicine of today — but not in the way most people believe. The public imagination remains fixated on "genetically modified babies" (the He Jiankui episode, 2018) or fantasies of genetic enhancement. The clinical reality is more mundane and more radical: CRISPR has already saved lives in patients with sickle cell disease, and it has just proven it can be mobilized in six months to save an infant unique in the world. The team demonstrated that with tight coordination between lab, LNP manufacturers, CROs, and regulators, a process that normally takes a decade can be compressed into 180 days. If this chain becomes replicable, it will transform the treatment of ultra-rare diseases. The question is no longer technical. It's economic, regulatory, and organizational.
Primary source
Musunuru K, Ahrens-Nicklas R, et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. New England Journal of Medicine. 2025. doi:10.1056/NEJMoa2504747. Project led by Children's Hospital of Philadelphia and Penn Medicine with philanthropic partners.
Key references
[1]Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829 — Founding paper of programmable CRISPR-Cas9.