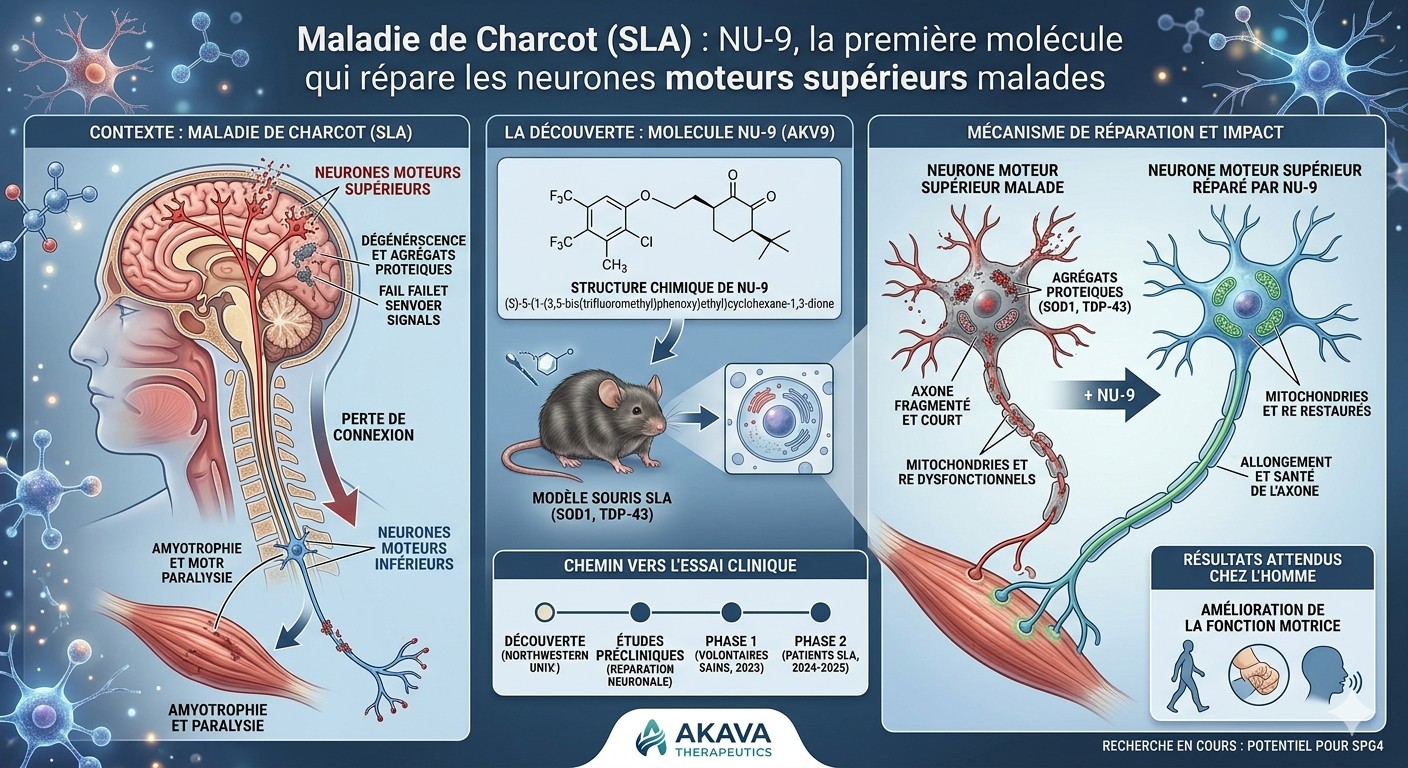
Een team van Northwestern University heeft een klein molecule, NU-9, geïdentificeerd dat de degeneratie kan stoppen van hersenneuronen die afsterven bij ALS — een primeur in de geschiedenis van deze ongeneeslijke ziekte.
Wat is ALS?
ALS (amyotrofe laterale sclerose), ook bekend als de ziekte van Charcot of de ziekte van Lou Gehrig, is een neurodegeneratieve aandoening waarbij motorneuronen geleidelijk afsterven. Deze neuronen vormen de bevelketen voor vrijwillige bewegingen: lopen, spreken, slikken, ademen. Wanneer ze verdwijnen, verzwakken de spieren en raken ze verlamd. De ziekte is momenteel ongeneeslijk, en de gemiddelde levensverwachting na diagnose is 3 tot 5 jaar.
Twee soorten motorneuronen, slechts één tot nu toe geviseerd
Motorneuronen zijn in twee niveaus georganiseerd. Bovenste motorneuronen ontspringen in de hersenen (motorische cortex) en lopen naar het ruggenmerg. Onderste motorneuronen vertrekken vanuit het ruggenmerg naar de spieren. Beide sterven af bij ALS — maar onderzoekers hebben zich historisch gericht op de onderste, die toegankelijker zijn. De bovenste werden als "secundair" beschouwd. Toch zijn het in sommige gevallen juist de bovenste die eerst degenereren.
Wat NU-9 in deze studie heeft gedaan
De onderzoekers werkten met twee muizenmodellen die ALS ontwikkelen: één door een defect SOD1-eiwit, het andere door het TDP-43-eiwit, dat bij ongeveer 95 % van de menselijke gevallen betrokken is. Ze gaven NU-9 oraal, dagelijks gedurende 60 dagen, vanaf het moment dat de muizen symptomen vertoonden.
Het resultaat, in cijfers
5
Zonder behandeling
bovenste motorneuronen behouden (op ~60 bij gezonde muis)
46
Met NU-9
neuronen behouden, bijna volledig herstel
Zonder behandeling hebben zieke muizen na 4 maanden nog maar 5 bovenste motorneuronen per geobserveerde doorsnede — in plaats van ongeveer 60 bij een gezonde muis. Met NU-9 behouden ze er 46. De molecule vertraagt de ziekte niet alleen: ze stopt de lopende degeneratie. De muizen bewegen ook beter: ze houden zich langer vast wanneer ze omgekeerd op een rooster worden gelegd, een test die specifiek de functie van neuronen in de motorische cortex meet.
Hoe doet NU-9 dat?
De molecule pakt twee problemen aan die zieke neuronen gemeen hebben: hun mitochondriën (de energiecentrales van de cel) zijn beschadigd, en hun endoplasmatisch reticulum (de fabriek waar eiwitten worden gemaakt) is verwijd en gefragmenteerd. NU-9 herstelt de structuur van beide. Concreet zien neuronen van behandelde muizen er onder de elektronenmicroscoop weer uit als gezonde neuronen.
⚠️ Wat dit NIET betekent
Deze studie werd uitgevoerd bij muizen, niet bij mensen. NU-9 beschermt hersenneuronen, maar niet die in het ruggenmerg (de onderste motorneuronen), die ook afsterven bij ALS. Veel moleculen die werken bij muizen falen bij mensen. Dit is dus geen beschikbare behandeling vandaag.
En sinds 2021?
Deze fundamentele studie werd gepubliceerd in 2021. Sindsdien werd NU-9 in licentie gegeven aan een biotechbedrijf, Akava Therapeutics, dat het hernoemde tot AKV9. De FDA gaf in augustus 2023 toestemming voor klinische studies bij mensen. Een Fase 1-studie startte in 2024 bij gezonde vrijwilligers om de tolerantie te beoordelen. In 2025 werden voorlopige gegevens over de ziekte van Alzheimer gepubliceerd in PNAS. Als de studies vorderen, kan een studie bij ALS-patiënten volgen. Het blijft een lange weg — het duurt vaak 10 tot 12 jaar voor een molecule van het laboratorium naar de apotheek gaat.
Wil je het volgende artikel rechtstreeks in je inbox?
Gratis · Geen spam · Volgend artikel deze week.
📌 De essentie in één zin
NU-9, een klein molecule oraal toegediend aan muizen met ALS, herstelt de integriteit van mitochondriën en endoplasmatisch reticulum in bovenste motorneuronen en stopt hun degeneratie in twee verschillende muizenmodellen (mSOD1 en TDP-43).
Waarom bovenste motorneuronen viseren?
Bovenste motorneuronen (UMN's, upper motor neurons) zijn de corticospinale neuronen van de motorische cortex — Betz-cellen in laag V. Ze verbinden de hersenen met het ruggenmerg en orkestreren vrijwillige beweging. Hun degeneratie is een diagnostische marker voor ALS, primaire laterale sclerose (PLS) en erfelijke spastische paraplegie (HSP). Toch werd op het moment van deze studie geen enkele molecule in klinische studies voor ALS getest op haar effect op UMN's. Alle huidige medicijnen (riluzol, edaravone) richten zich indirect of voornamelijk op onderste motorneuronen en de bijbehorende neuro-inflammatie.
Twee muizenmodellen, twee verschillende oorzaken
ALS heeft geen enkele oorzaak. De onderzoekers testten NU-9 in twee muizenmodellen die twee onafhankelijke, niet-overlappende mechanismen vertegenwoordigen:
hSOD1G93A-UeGFP: brengt gemuteerd menselijk SOD1-eiwit tot overexpressie. Modelleert de toxiciteit van verkeerd gevouwen SOD1 (familiale vormen).
prpTDP-43A315T-UeGFP: brengt gemuteerd TDP-43-eiwit tot overexpressie. Modelleert TDP-43-pathologie, aanwezig bij ongeveer 95 % van de ALS-patiënten.
Belangrijke nuance: deze twee pathologieën komen zelden samen voor. Patiënten met een SOD1-mutatie hebben geen TDP-43-aggregaten, en omgekeerd. Een molecule vinden die in beide situaties werkt suggereert dat ze inwerkt op een gedeeld cellulair mechanisme, downstream van de aansturende eiwitten.
Het protocol in het kort
Bovenste motorneuronen werden gelabeld met groene fluorescentie (GFP) via een transgen (UCHL1-eGFP), waardoor ze rechtstreeks onder de microscoop te identificeren zijn. NU-9 werd dagelijks toegediend via orale sondevoeding van postnatale dag 60 (P60, begin van symptomen) tot P120 (gevorderd stadium), in twee doses: 20 of 100 mg/kg/dag. De controlegroep kreeg het oplosmiddel (olijfolie + NMP).
Belangrijkste resultaten
Parameter (per doorsnede)
Gezonde muis
Onbehandeld ALS-model
ALS + NU-9 (100 mg/kg/d)
mSOD1-model
UMN's behouden
59 ± 3
5 ± 2
46 ± 3
Gezonde mitochondriën
93 %
10 %
86 %
Apicale dendrieten met vacuoles
28 %
76 %
23 %
TDP-43-model
UMN's behouden
59 ± 4
37 ± 4
59 ± 3
Gezonde mitochondriën
93 %
22 %
87 %
Apicale dendrieten met vacuoles
33 %
81 %
11 %
Alle verschillen tussen behandelde en onbehandelde groepen zijn statistisch significant (p<0,01 tot p<0,0001 afhankelijk van de parameter). In het TDP-43-model is het herstel bijna volledig: behandelde muizen zijn niet te onderscheiden van gezonde controles op de gemeten cellulaire parameters.
Effect op motorisch gedrag
De hanging wire-test, bijzonder gevoelig voor UMN-functie, toont een significante verbetering bij behandelde muizen vanaf P102 (mSOD1) of P120 (TDP-43), met een terugkeer naar prestaties van gezonde muizen. De rotarodtest, algemener, toont alleen verbetering in het TDP-43-model.
⚠️ Grote beperking: geen effect op onderste motorneuronen
In het ruggenmerg blijven onderste motorneuronen (LMN's) in het mSOD1-model degenereren ondanks de behandeling. NU-9 redt niet alles — het werkt specifiek op UMN's van de motorische cortex. Voor menselijke ALS, waar beide populaties afsterven, zal NU-9 alleen waarschijnlijk niet volstaan. Een combinatie met andere geneesmiddelen (riluzol, edaravone) wordt in latere publicaties onderzocht.
Voorgesteld mechanisme
NU-9 werd aanvankelijk geïdentificeerd voor zijn vermogen om de aggregatie van gemuteerd SOD1 in celkweken te verminderen. Het verlaagt inderdaad het niveau van verkeerd gevouwen SOD1 in behandelde UMN's (~–28 %). Maar zijn werkzaamheid in het TDP-43-model, waar geen gemuteerd SOD1 is, wijst op een breder mechanisme: de molecule herstelt de gedeelde organellen (mitochondriën, ER) waarvan de disfunctie door beide pathologieën gedeeld wordt. Het precieze moleculaire mechanisme is gedeeltelijk gekarakteriseerd — latere publicaties (2025) wijzen op een lysosomale route.
Van labo naar mens: het vervolg
Deze studie uit 2021 legde de basis. Het vervolg is concreet: NU-9 werd in licentie gegeven aan Akava Therapeutics (opgericht door Richard Silverman, de uitvinder van de molecule, die ook pregabaline / Lyrica heeft ontworpen). De FDA verleende in augustus 2023 IND-clearance en gaf groen licht voor klinische studies. Akava hernoemde de molecule tot AKV9. Een Fase 1-studie bij gezonde vrijwilligers beoordeelt verdraagbaarheid en farmacokinetiek. In 2025 toonden resultaten in PNAS een positief effect van AKV9 in modellen van de ziekte van Alzheimer, wat wijst op een gedeeld mechanisme bij neurodegeneratieve ziekten.
Genç et al. (Clin Transl Med 2021;11:e336, DOI: 10.1002/ctm2.336) tonen aan dat een chiraal cyclohexaan-1,3-dion-derivaat (NU-9, 100 mg/kg/d PO, P60→P120) de mitochondriale ultrastructuur (binnenmembraan, cristae) en het ruwe ER (cisternae, ribosomen) herstelt, mSOD1-aggregatie vermindert, apicale dendrieten stabiliseert en UMN-degeneratie stopt in hSOD1G93A- en prpTDP-43A315T-muizenmodellen, zonder effect op spinale LMN's.
Oorsprong en chemie van NU-9
NU-9 komt voort uit een high-throughput screening van meer dan 50 000 verbindingen (Silverman Lab, Northwestern) in een duale SOD1G93A-PC12-celassay: bescherming tegen cytotoxiciteit en remming van aggregatie. Initiële hits werden gefilterd via computationele benaderingen (substructuuronderzoek, ligand-based clustering) en vervolgens via medicinale chemie geoptimaliseerd. Hit-verbinding 1, uit de cyclohexaan-1,3-dion-familie, leidde tot verbinding 2 (goede ADME maar slechte neuronale penetratie), en vervolgens tot NU-9, een chiraal derivaat met een tert-butylgroep en een bis-trifluormethyl-fenylgroep.
ADME / farmacokinetisch profiel
Parameter
Waarde
EC50 (aggregatie/toxiciteit)
300 nM
Wateroplosbaarheid
≥100 µM
Microsoomstabiliteit (t½)
74 min (mens) / 52 min (muis)
Plasma-eiwitbinding
90 %
PAMPA-BBB
8,11 × 10⁻⁶ cm/s (CNS+)
Hersenpenetratie
8,3 µM
Orale biobeschikbaarheid
94 %
t½ in vivo (muis)
2,73 h
hERG / CYP-remming (5)
Geen (tot 30 µM) / <10 % bij 3 µM
NOAEL (muis, PO, 7 d)
100 mg/kg
Levensduurverlenging (mSOD1)
+13 % bij 20 mg/kg
Methodologie: de UCHL1-eGFP-lijn
Belangrijke methodologische innovatie: de UCHL1-eGFP-lijn (Ozdinler Lab, JAX 022476) labelt selectief UMN's met stabiele GFP. Gekruist met hSOD1G93A of prpTDP-43A315T maakt ze het mogelijk om in vivo zieke UMN's te identificeren, essentieel om de cellulaire effecten van een verbinding te kwantificeren. Dit is het eerste preklinische platform dat het mogelijk maakt de UMN-respons op behandeling te beoordelen.
Elektronenmicroscopie: wat er in de cel gebeurt
Ultrastructurele analyse via TEM (FEI Tecnai Spirit G2) op ultradunne coupes (70 nm) van de motorische cortex onthult de karakteristieke afwijkingen van zieke UMN's: mitochondriën met gedesintegreerd binnenmembraan, verlies van cristae, zwelling van cisternae van het ruwe ER, ribosomenfragmentatie. Deze afwijkingen zijn behouden tussen muizenmodellen en menselijke Betz-cellen (post-mortemstalen van 9 ALS-patiënten en 4 controles, Northwestern Brain Bank), wat de translationele relevantie bevestigt.
De afwezigheid van effect op LMN's in het mSOD1-model is verwacht en informatief: bij beide geteste doses verschilt het aantal kwetsbare ChAT⁺- of NeuN⁺/ChAT⁺-LMN's niet van vehiculum (~10–14/doorsnede vs ~46/doorsnede bij WT). Deze specificiteit pleit voor een UMN-specifiek cellulair doel — mogelijk gerelateerd aan hun morfologische bijzonderheden (zeer lange axonen, dichte corticale integratie, aanhoudende energievraag) of hun differentieel transcriptoom. In het TDP-43-model is er geen significant LMN-verlies op P120, wat beoordeling in deze populatie verhindert.
Kritische analyse en beperkingen
Preklinische muizenstudie: extrapolatie naar de mens blijft hypothetisch. Het faalpercentage bij overgang van ALS-muizen naar klinische studies is historisch hoog (riluzol is de uitzondering die de regel bevestigt).
Bescheiden TDP-43-aantallen: n=4 behandelde muizen vs n=3 controles. Robuust voor ultrastructurele effecten, maar beperkt voor mortaliteit of subtielere effecten.
Gedeeltelijke muizenmodellen: TDP-43-pathologie geïnduceerd door overexpressie van een gemuteerd transgen (A315T) reproduceert niet exact de progressieve cytoplasmatische dislocatie die bij de meerderheid van patiënten wordt waargenomen (geaggregeerd wild-type TDP-43).
Onnauwkeurig moleculair mechanisme: op het moment van deze publicatie was het directe doelwit van NU-9 niet geïdentificeerd. Latere publicaties (PNAS 2025) wijzen op een lysosoomafhankelijk mechanisme van eiwitaggregatie-remming.
Niet alle gedragsanalyses waren geblindeerd: de experimentatoren waren op de hoogte van de conditie (behandeld/onbehandeld) tijdens rotarod- en hanging wire-tests. Cellulaire analyses werden geblindeerd uitgevoerd.
Negatief rotarod-effect in mSOD1-model: contrast met positief effect op hanging wire. De differentiële gevoeligheid van deze tests voor UMN-functie verklaart dit resultaat waarschijnlijk, maar het beperkt de veralgemening van gedragsmatige voordelen.
Traject 2021–2026
Feb. 2021: publicatie van de fundamentele studie (Genç et al., Clin Transl Med).
Maart 2022: Genç et al. (Sci Rep) — NU-9 overtreft riluzol en edaravone alleen, en verhoogt hun werkzaamheid in combinatie op UMN-axonregeneratie.
Juni 2022: Akava Therapeutics neemt NU-9 (Northwestern) in licentie; hernoemd tot AKV9.
Juli 2022: Orphan Drug Designation verkregen van de FDA (sectie 526 FD&C).
Aug. 2023: FDA IND-clearance voor AKV9 in ALS. Fase 1 bij gezonde vrijwilligers toegestaan (single + multiple ascending doses).
Dec. 2024: subsidie van 7,3 M$ van het National Institute on Aging (NIH) om AKV9 uit te breiden naar neurodegeneratieve ziekten.
Mei 2025: PNAS-publicatie over AKV9 en de ziekte van Alzheimer (PMID 40030015) — 3 op 4 behandelde muizen behouden een normaal geheugen in een proof-of-concept-studie.
⚠️ Belangenconflicten
Op het moment van publicatie (2021) verklaarden de auteurs geen belangenconflicten. Sinds 2022 is R.B. Silverman oprichter van Akava Therapeutics, dat AKV9 commercialiseert; Northwestern University heeft financiële belangen (royalty's, aandelen). P.H. Özdinler is SAB Chair van Akava. Deze affiliaties moeten worden meegenomen bij de interpretatie van latere publicaties van de groep.
Implicaties en vooruitzichten
Deze studie introduceert twee belangrijke concepten voor de ontwikkeling van therapieën voor motorneuronaandoeningen: (1) UMN-gezondheid moet een afzonderlijk preklinisch eindpunt zijn, wat nooit eerder werd gedaan voor verbindingen in klinische studies; (2) mitochondriale en ER-defecten convergeren stroomafwaarts van heterogene oorzaken (mSOD1, TDP-43, mogelijk C9orf72 en andere), wat een gedeeld farmacologisch doelwit vormt. De potentiële reikwijdte overstijgt ALS — PLS, HSP, ALS/FTLD, en nu ook Alzheimer. Het blijft af te wachten of Fase 1 van AKV9, en latere fases, een werkzaamheidssignaal bij mensen zullen bevestigen.
Genç B, Gautam M, Gözütok Ö, et al. Improving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology. Clin Transl Med. 2021;11(2):e336. doi:10.1002/ctm2.336. PMID: 33634973.
Veelgestelde vragen
Wanneer zal NU-9 (AKV9) beschikbaar zijn voor ALS-patiënten?
Niet voor verschillende jaren, en alleen als de studies slagen. De FDA gaf in augustus 2023 toestemming voor Fase 1 bij gezonde vrijwilligers, die verdraagbaarheid en farmacokinetiek beoordeelt. Een Fase 2 bij ALS-patiënten kan volgen, daarna een bredere Fase 3 voor enige commercialisatie. De gebruikelijke vertraging tussen de eerste studies bij mensen en marktautorisatie is 10 tot 12 jaar volgens Richard Silverman, de uitvinder van de molecule. Een realistische beschikbaarheid zou rond 2030–2035 liggen in het meest gunstige scenario.
Kan NU-9 ALS genezen?
De studie suggereert geen genezing, zelfs niet bij muizen. NU-9 stopt de degeneratie van bovenste motorneuronen (hersenschors), maar heeft geen aangetoond effect op onderste motorneuronen (ruggenmerg), die ook afsterven bij ALS. Een effectieve therapie zou NU-9 waarschijnlijk moeten combineren met andere geneesmiddelen die onderste motorneuronen viseren, zoals riluzol of edaravone. Latere publicaties van dezelfde groep (Sci Rep, 2022) hebben deze combinatiebenadering onderzocht.
Zijn NU-9 en AKV9 dezelfde molecule?
Ja. NU-9 is de academische naam gegeven door de Northwestern University-laboratoria. In juni 2022 werd de molecule in licentie gegeven aan Akava Therapeutics, een biotechbedrijf opgericht door Richard Silverman zelf, die de molecule hernoemde tot AKV9 voor de klinische ontwikkeling. Het is exact dezelfde chemische verbinding.
Waarom werd deze studie alleen bij muizen uitgevoerd?
Het is de verplichte preklinische stap voorafgaand aan elke studie bij mensen. De gebruikte muizenmodellen (hSOD1G93A en prpTDP-43A315T) werden gekozen omdat ze de cellulaire veranderingen in bovenste motorneuronen van ALS-patiënten getrouw reproduceren — geverifieerd op menselijke post-mortemstalen in dezelfde studie. De overgang naar de mens vereist aanvullende veiligheidsstudies (toxicologie, farmacokinetiek bij grote dieren), wat Akava Therapeutics ondernam voor de FDA IND-clearance in 2023.
Hoe verschilt deze studie van eerdere ALS-klinische studies?
Dit is de eerste studie die expliciet de gezondheid van bovenste motorneuronen als preklinisch criterium evalueert. Geneesmiddelen in ontwikkeling voor ALS hebben historisch verlenging van de levensduur van de muis of overleving van onderste motorneuronen als belangrijkste indicator gebruikt. Veel van deze moleculen zijn echter gefaald bij mensen. Genç et al. stellen een paradigmaverschuiving voor: zich richten op de neuronen die werkelijk (en vroeg) degenereren bij patiënten, en cellulaire indicatoren gebruiken (mitochondriën, ER, dendrieten) in plaats van gedrags- of overlevingsindicatoren.
Als NU-9 onderste motorneuronen niet redt, wat is dan het nut?
Ten eerste om de hypothese te valideren dat het mogelijk is om de degeneratie van één type motorneuron in ALS te stoppen — wat nooit was aangetoond. Ten tweede omdat bovenste motorneuronen een cruciale rol spelen in vrijwillige motorische controle, en betrokken zijn bij verschillende andere ziekten (primaire laterale sclerose, erfelijke spastische paraplegie, ALS/FTLD). Ten slotte, in een gecombineerde therapeutische strategie: NU-9 zou UMN's beschermen terwijl andere moleculen LMN's viseren. Latere gegevens (2022) tonen zelfs aan dat NU-9 de werkzaamheid van riluzol en edaravone op axonregeneratie verhoogt.
Kan NU-9 effectief zijn bij andere neurodegeneratieve ziekten?
Mogelijk. De wetenschappelijke logica is solide: mitochondriale disfunctie en eiwitaggregatie zijn mechanismen gedeeld door veel neurodegeneratieve ziekten, waaronder de ziekte van Alzheimer, de ziekte van Parkinson, frontotemporale degeneratie (FTLD) en de ziekte van Huntington. In mei 2025 toonde een publicatie in PNAS aan dat AKV9 het geheugen verbetert in een muizenmodel van de ziekte van Alzheimer (3 op 4 behandelde muizen herwinnen een normaal geheugen). Akava Therapeutics verkreeg ook een financiering van 7,3 M$ van het National Institute on Aging in 2024 om het onderzoek uit te breiden buiten ALS.
🍪 Deze site gebruikt Google Analytics en de Facebook Pixel om het publiek te meten. Deze tools worden alleen geactiveerd met uw toestemming. Privacybeleid →