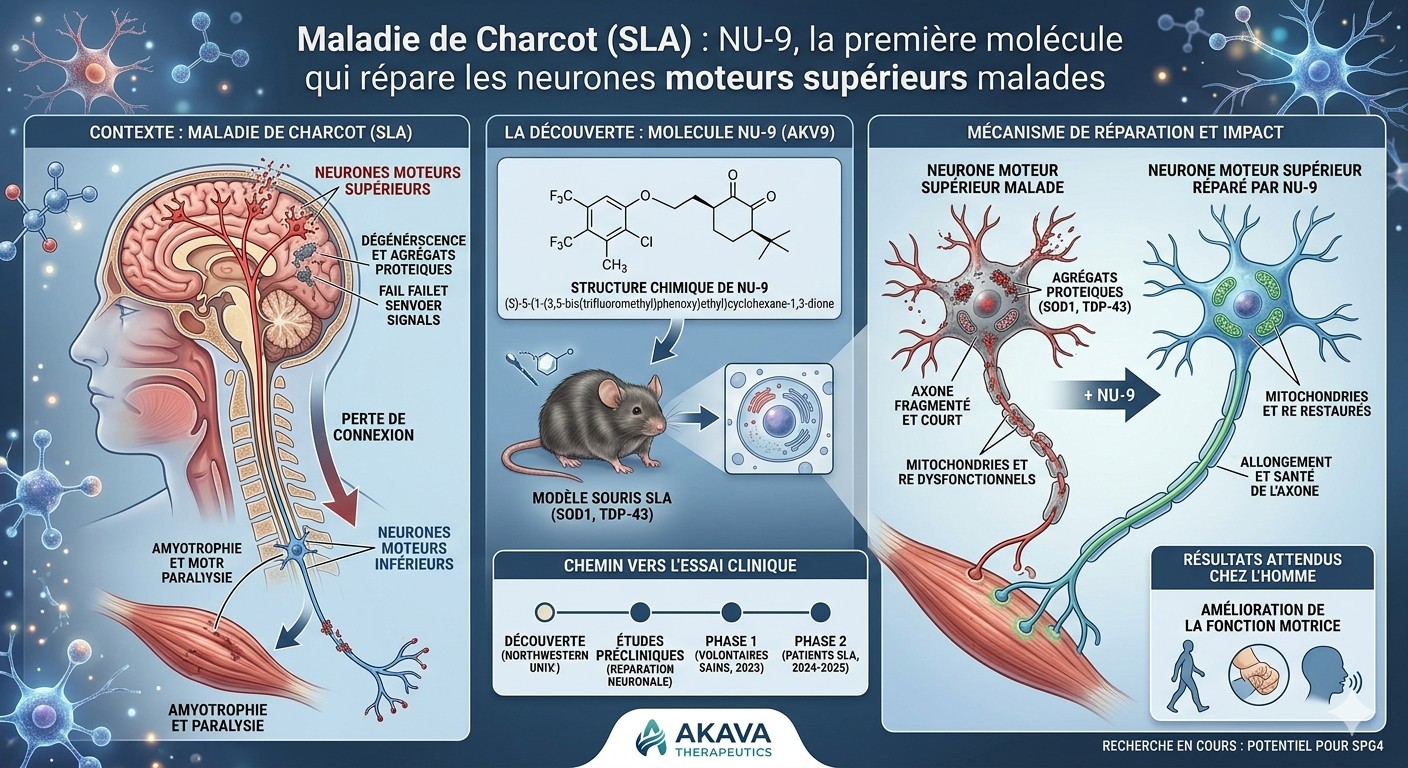
Une équipe de Northwestern University a identifié une petite molécule, NU-9, capable de stopper la dégénérescence des neurones du cerveau qui meurent dans la maladie de Charcot — une première dans l'histoire de cette maladie incurable.
C'est quoi la maladie de Charcot ?
La maladie de Charcot, aussi appelée SLA (sclérose latérale amyotrophique), est une maladie neurodégénérative dans laquelle les neurones moteurs meurent progressivement. Ces neurones sont la chaîne de commande des mouvements volontaires : marcher, parler, avaler, respirer. Quand ils disparaissent, les muscles s'affaiblissent, puis se paralysent. La maladie est aujourd'hui incurable, et l'espérance de vie moyenne après diagnostic est de 3 à 5 ans.
Deux types de neurones moteurs, un seul ciblé jusqu'ici
Les neurones moteurs sont organisés en deux étages. Les neurones moteurs supérieurs partent du cerveau (cortex moteur) et descendent dans la moelle épinière. Les neurones moteurs inférieurs, eux, partent de la moelle épinière et atteignent les muscles. Les deux meurent dans la SLA — mais les chercheurs se sont historiquement concentrés sur les inférieurs, plus accessibles. Les supérieurs étaient considérés comme « secondaires ». Pourtant, ce sont eux qui dégénèrent en premier dans certains cas.
Ce que NU-9 a fait dans cette étude
Les chercheurs ont travaillé sur deux modèles de souris qui développent une SLA : l'un à cause d'une protéine SOD1 défectueuse, l'autre à cause de la protéine TDP-43, qui est impliquée dans environ 95 % des cas humains. Ils ont donné NU-9 par voie orale tous les jours pendant 60 jours, à partir du moment où les souris commencent à montrer des symptômes.
Le résultat, chiffré
5
Sans traitement
neurones moteurs supérieurs préservés (sur ~60 chez la souris saine)
46
Avec NU-9
neurones préservés, soit un retour quasi-normal
Sans traitement, à 4 mois, les souris malades n'ont plus que 5 neurones moteurs supérieurs par section observée — au lieu d'environ 60 chez une souris saine. Avec NU-9, elles en conservent 46. La molécule ne se contente pas de ralentir la maladie : elle arrête la dégénérescence en cours. Les souris bougent aussi mieux : elles se cramponnent plus longtemps lorsqu'on les retourne sur une grille, un test qui mesure spécifiquement la fonction des neurones du cortex moteur.
Comment NU-9 fait-il ça ?
La molécule s'attaque à deux problèmes communs aux neurones malades : leurs mitochondries (les centrales énergétiques de la cellule) sont cassées, et leur réticulum endoplasmique (l'usine où sont fabriquées les protéines) est dilaté et fragmenté. NU-9 restaure la structure des deux. Concrètement, en regardant les neurones au microscope électronique, ceux des souris traitées ressemblent à nouveau à des neurones sains.
⚠️ Ce que ça ne veut PAS dire
Cette étude a été faite chez la souris, pas chez l'humain. NU-9 protège les neurones du cerveau, mais pas ceux de la moelle épinière (les neurones moteurs inférieurs), qui meurent aussi dans la SLA. Beaucoup de molécules qui marchent chez la souris échouent chez l'humain. Ce n'est donc pas un traitement disponible aujourd'hui.
Et depuis 2021 ?
Cette étude fondatrice a été publiée en 2021. Depuis, NU-9 a été licencié à une société de biotech, Akava Therapeutics, qui l'a renommé AKV9. La FDA a autorisé en août 2023 le passage à des essais cliniques sur l'humain. Une étude de Phase 1 a démarré en 2024 chez des volontaires sains pour vérifier la tolérance. En 2025, des données préliminaires sur la maladie d'Alzheimer ont été publiées dans PNAS. Si les essais avancent, un essai sur des patients SLA pourrait suivre. Ça reste long — il faut souvent 10 à 12 ans pour qu'une molécule passe du laboratoire à la pharmacie.
Tu veux le prochain article directement dans ta boîte mail ?
Gratuit · Zéro spam · Le prochain article sort cette semaine.
📌 L'essentiel en une phrase
NU-9, une petite molécule administrée par voie orale à des souris atteintes de SLA, restaure l'intégrité des mitochondries et du réticulum endoplasmique des neurones moteurs supérieurs et stoppe leur dégénérescence dans deux modèles murins distincts (mSOD1 et TDP-43).
Pourquoi cibler les neurones moteurs supérieurs ?
Les neurones moteurs supérieurs (UMN, upper motor neurons) sont les neurones corticospinaux du cortex moteur — les cellules de Betz dans la couche V. Ils relient le cerveau à la moelle épinière et orchestrent le mouvement volontaire. Leur dégénérescence est un marqueur diagnostique de la SLA, de la sclérose latérale primitive (SLP) et de la paraplégie spastique héréditaire (PSH). Pourtant, à la date de cette étude, aucune molécule en essai clinique pour la SLA n'avait été testée pour son effet sur les UMN. Tous les médicaments actuels (riluzole, edaravone) ciblent indirectement ou prioritairement les motoneurones inférieurs et la neuro-inflammation associée.
Deux modèles murins, deux causes distinctes
La SLA n'a pas une cause unique. Les chercheurs ont testé NU-9 dans deux modèles murins représentant deux mécanismes indépendants et non chevauchants :
hSOD1G93A-UeGFP : surexprime la protéine SOD1 humaine mutée. Modélise la toxicité de la SOD1 mal repliée (formes familiales).
prpTDP-43A315T-UeGFP : surexprime la protéine TDP-43 mutée. Modélise la pathologie TDP-43, présente chez environ 95 % des patients SLA.
La nuance importante : ces deux pathologies coexistent rarement. Les patients avec mutation SOD1 n'ont pas d'agrégats TDP-43, et inversement. Trouver une molécule efficace dans les deux situations suggère qu'elle agit sur un mécanisme cellulaire commun, en aval des protéines déclenchantes.
Le protocole en bref
Les neurones moteurs supérieurs ont été marqués par fluorescence verte (GFP) grâce à un transgène (UCHL1-eGFP), permettant de les identifier directement au microscope. NU-9 a été administré quotidiennement par gavage oral du jour postnatal 60 (P60, début des symptômes) au jour P120 (stade avancé), à deux doses : 20 ou 100 mg/kg/jour. Les contrôles ont reçu le véhicule (huile d'olive + NMP).
Résultats principaux
Paramètre (par section)
Souris saine
Modèle SLA non traité
SLA + NU-9 (100 mg/kg/j)
Modèle mSOD1
UMN préservés
59 ± 3
5 ± 2
46 ± 3
Mitochondries saines
93 %
10 %
86 %
Dendrites apicales avec vacuoles
28 %
76 %
23 %
Modèle TDP-43
UMN préservés
59 ± 4
37 ± 4
59 ± 3
Mitochondries saines
93 %
22 %
87 %
Dendrites apicales avec vacuoles
33 %
81 %
11 %
Tous les écarts entre groupes traités et non traités sont statistiquement significatifs (p<0,01 à p<0,0001 selon les paramètres). Dans le modèle TDP-43, le retour est presque complet : les souris traitées sont indiscernables des contrôles sains sur les paramètres cellulaires mesurés.
Effet sur le comportement moteur
Le test de la grille suspendue (hanging wire test), particulièrement sensible à la fonction des UMN, montre une amélioration significative chez les souris traitées dès P102 (mSOD1) ou P120 (TDP-43), avec un retour aux performances des souris saines. Le test du rotarod, plus généraliste, montre une amélioration uniquement dans le modèle TDP-43.
⚠️ Limite majeure : pas d'effet sur les motoneurones inférieurs
Dans la moelle épinière, les motoneurones inférieurs (LMN) du modèle mSOD1 continuent de dégénérer malgré le traitement. NU-9 ne sauve pas tout — il agit spécifiquement sur les UMN du cortex moteur. Pour une SLA humaine, où les deux populations meurent, NU-9 seul ne sera probablement pas suffisant. Une combinaison avec d'autres molécules (riluzole, edaravone) est explorée dans des publications ultérieures.
Mécanisme proposé
NU-9 a été initialement identifié pour sa capacité à réduire l'agrégation de la SOD1 mutée dans des cellules en culture. Il diminue effectivement les niveaux de SOD1 mal repliée dans les UMN traités (–28 % environ). Mais son efficacité dans le modèle TDP-43, où il n'y a pas de SOD1 mutée, suggère un mécanisme plus large : la molécule restaure les organites communs (mitochondries, RE) dont la défaillance est partagée par les deux pathologies. Le mécanisme moléculaire précis reste partiellement caractérisé — des publications ultérieures (2025) suggèrent une voie lysosomale.
Du laboratoire à l'humain : la suite
Cette étude de 2021 a posé les bases. La suite est concrète : NU-9 a été licencié à Akava Therapeutics (fondée par Richard Silverman, l'inventeur de la molécule, qui avait également conçu la prégabaline / Lyrica). La FDA a accordé l'IND clearance en août 2023, autorisant les essais cliniques. Akava a renommé la molécule AKV9. Une étude de Phase 1 chez des volontaires sains évalue sa tolérance et sa pharmacocinétique. En 2025, des résultats publiés dans PNAS ont montré un effet positif d'AKV9 dans des modèles de maladie d'Alzheimer, suggérant un mécanisme commun aux maladies neurodégénératives.
Genç et al. (Clin Transl Med 2021;11:e336, DOI : 10.1002/ctm2.336) démontrent qu'un dérivé chiral de cyclohexane-1,3-dione (NU-9, 100 mg/kg/j PO, P60→P120) restaure l'ultrastructure mitochondriale (membrane interne, crêtes) et du RE rugueux (citernes, ribosomes), réduit l'agrégation de mSOD1, stabilise les dendrites apicales et stoppe la dégénérescence des UMN dans les modèles murins hSOD1G93A et prpTDP-43A315T, sans effet sur les LMN spinaux.
Origine et chimie de NU-9
NU-9 est issu d'un criblage haut-débit de plus de 50 000 composés (laboratoire Silverman, Northwestern) dans un essai cellulaire SOD1G93A-PC12 dual : protection contre la cytotoxicité et inhibition de l'agrégation. Les hits initiaux ont été filtrés par approches computationnelles (recherche de sous-structures, clustering ligand-based) puis optimisés par chimie médicinale en plusieurs cycles. Le composé hit 1, de la famille cyclohexane-1,3-dione, a donné le composé 2 (bonne ADME mais faible pénétration neuronale), puis NU-9, un dérivé chiral avec ajout d'un groupement tert-butyle et d'un groupement bis-trifluorométhyl-phényle.
Profil ADME / pharmacocinétique
Paramètre
Valeur
EC50 (agrégation/toxicité)
300 nM
Solubilité aqueuse
≥100 µM
Stabilité microsomes (t½)
74 min (humain) / 52 min (souris)
Liaison aux protéines plasmatiques
90 %
PAMPA-BBB
8,11 × 10⁻⁶ cm/s (CNS+)
Pénétration cérébrale
8,3 µM
Biodisponibilité orale
94 %
t½ in vivo (souris)
2,73 h
hERG / inhibition CYP (5)
Aucune (jusqu'à 30 µM) / <10 % à 3 µM
NOAEL (souris, PO, 7 j)
100 mg/kg
Extension de survie (mSOD1)
+13 % à 20 mg/kg
Méthodologie : la lignée UCHL1-eGFP
Innovation méthodologique majeure : la lignée UCHL1-eGFP (Ozdinler Lab, JAX 022476) marque sélectivement les UMN par GFP stable. Croisée avec hSOD1G93A ou prpTDP-43A315T, elle permet d'identifier in vivo les UMN malades, indispensable pour quantifier les effets cellulaires d'un composé. C'est la première plateforme préclinique permettant d'évaluer la réponse des UMN à un traitement.
Microscopie électronique : ce qui se passe dans la cellule
L'analyse ultrastructurale par TEM (FEI Tecnai Spirit G2) sur coupes ultrafines (70 nm) de cortex moteur révèle les anomalies caractéristiques des UMN malades : mitochondries avec membrane interne désintégrée, perte des crêtes, gonflement des citernes du RE rugueux, fragmentation des ribosomes. Ces anomalies sont conservées entre les modèles murins et les cellules de Betz humaines (échantillons post-mortem de 9 patients SLA et 4 contrôles, Northwestern Brain Bank), validant la pertinence translationnelle.
L'absence d'effet sur les LMN du modèle mSOD1 est attendue et instructive : aux deux doses testées, le nombre de LMN ChAT⁺ ou NeuN⁺/ChAT⁺ vulnérables ne diffère pas du véhicule (~10–14/section vs ~46/section chez WT). Cette spécificité plaide pour une cible cellulaire propre aux UMN — possiblement liée à leurs particularités morphologiques (axones très longs, intégration corticale dense, demande énergétique soutenue) ou à leur transcriptome différentiel. Dans le modèle TDP-43, il n'y a pas de perte significative de LMN à P120, ce qui empêche d'évaluer le composé sur cette population.
Analyse critique et limites
Étude préclinique murine : l'extrapolation à l'humain reste hypothétique. Le taux d'échec en passant de la souris SLA aux essais cliniques est historiquement élevé (riluzole étant l'exception qui confirme la règle).
Effectifs TDP-43 modestes : n=4 souris traitées vs n=3 contrôles. Robuste pour les effets ultrastructuraux, mais limité pour la mortalité ou des effets plus subtils.
Modèles murins partiels : la pathologie TDP-43 induite par surexpression d'un transgène mutant (A315T) ne reproduit pas exactement la délocalisation cytoplasmique progressive observée chez la majorité des patients (TDP-43 sauvage agrégé).
Mécanisme moléculaire imprécis : au moment de cette publication, la cible directe de NU-9 n'était pas identifiée. Des publications ultérieures (PNAS 2025) suggèrent un mécanisme lysosome-dépendant d'inhibition de l'agrégation protéique.
Pas d'analyse en aveugle de tous les paramètres comportementaux : les expérimentateurs étaient au courant de la condition (traité/non traité) lors des tests rotarod et hanging wire. Les analyses cellulaires ont été faites en aveugle.
Effet sur le rotarod négatif dans le modèle mSOD1 : contraste avec l'effet positif sur hanging wire. La sensibilité différentielle de ces tests à la fonction UMN explique probablement ce résultat, mais cela limite la généralisation des bénéfices comportementaux.
Trajectoire 2021–2026
Févr. 2021 : publication de l'étude fondatrice (Genç et al., Clin Transl Med).
Mars 2022 : Genç et al. (Sci Rep) — NU-9 surpasse riluzole et edaravone seuls, et augmente leur efficacité en combinaison sur la régénération axonale des UMN.
Juin 2022 : Akava Therapeutics licencie NU-9 (Northwestern) ; renommage en AKV9.
Juill. 2022 : Orphan Drug Designation obtenu auprès de la FDA (section 526 FD&C).
Août 2023 : FDA IND clearance pour AKV9 dans la SLA. Phase 1 chez volontaires sains autorisée (single + multiple ascending doses).
Déc. 2024 : grant de 7,3 M$ du National Institute on Aging (NIH) pour étendre AKV9 aux maladies neurodégénératives.
Mai 2025 : publication PNAS sur AKV9 et la maladie d'Alzheimer (PMID 40030015) — 3 souris traitées sur 4 conservent une mémoire normale dans une étude de proof-of-concept.
⚠️ Conflits d'intérêts
À la date de la publication (2021), les auteurs déclaraient une absence de conflit d'intérêts. Depuis 2022, R.B. Silverman est fondateur d'Akava Therapeutics qui commercialise AKV9 ; Northwestern University détient des intérêts financiers (royalties, equity). P.H. Özdinler est SAB Chair d'Akava. Ces affiliations sont à prendre en compte lors de l'interprétation des publications ultérieures du groupe.
Implications et perspectives
Cette étude introduit deux concepts importants pour le développement de thérapies dans les maladies des motoneurones : (1) la santé des UMN doit être un critère d'évaluation préclinique distinct, ce qui n'avait jamais été fait auparavant pour les composés en essai clinique ; (2) les défauts mitochondriaux et du RE convergent en aval de causes hétérogènes (mSOD1, TDP-43, possiblement C9orf72 et autres), constituant une cible pharmacologique partagée. La portée potentielle dépasse la SLA — SLP, PSH, ALS/FTLD, et désormais Alzheimer. Reste à voir si la Phase 1 d'AKV9, et les phases ultérieures, confirmeront un signal d'efficacité chez l'humain.
Genç B, Gautam M, Gözütok Ö, et al. Improving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology. Clin Transl Med. 2021;11(2):e336. doi:10.1002/ctm2.336. PMID: 33634973.
Questions fréquentes
Quand NU-9 (AKV9) sera-t-il disponible pour les patients atteints de SLA ?
Pas avant plusieurs années, et seulement si les essais réussissent. La FDA a autorisé en août 2023 la Phase 1 chez des volontaires sains, qui évalue la tolérance et la pharmacocinétique. Une Phase 2 chez des patients SLA pourrait suivre, puis une Phase 3 plus large avant toute commercialisation. Le délai habituel entre les premiers essais humains et la mise sur le marché d'un nouveau médicament est de 10 à 12 ans selon Richard Silverman, l'inventeur de la molécule. Une disponibilité réaliste se situerait autour de 2030–2035 dans le scénario le plus favorable.
NU-9 peut-il guérir la maladie de Charcot ?
L'étude ne suggère pas une guérison, même chez la souris. NU-9 stoppe la dégénérescence des neurones moteurs supérieurs (cortex cérébral), mais n'a aucun effet démontré sur les motoneurones inférieurs (moelle épinière), qui meurent aussi dans la SLA. Une éventuelle thérapie efficace devrait probablement combiner NU-9 avec d'autres molécules ciblant les motoneurones inférieurs, comme le riluzole ou l'edaravone. Des publications ultérieures du même groupe (Sci Rep, 2022) ont d'ailleurs exploré cette piste de combinaison.
NU-9 et AKV9 sont-ils la même molécule ?
Oui. NU-9 est le nom académique donné par les laboratoires de Northwestern University. En juin 2022, la molécule a été licenciée à Akava Therapeutics, une société de biotechnologie fondée par Richard Silverman lui-même, qui l'a renommée AKV9 dans le cadre de son développement clinique. C'est exactement le même composé chimique.
Pourquoi cette étude n'a-t-elle été faite que chez la souris ?
C'est l'étape préclinique obligatoire avant tout essai humain. Les modèles murins utilisés (hSOD1G93A et prpTDP-43A315T) ont été choisis car ils reproduisent fidèlement les altérations cellulaires observées dans les neurones moteurs supérieurs des patients SLA — vérifiées sur des échantillons post-mortem humains dans la même étude. Le passage à l'humain demande des études de sécurité supplémentaires (toxicologie, pharmacocinétique sur grand animal), ce qu'Akava Therapeutics a entrepris avant l'IND clearance de la FDA en 2023.
En quoi cette étude diffère-t-elle des essais cliniques précédents pour la SLA ?
C'est la première étude qui évalue explicitement la santé des neurones moteurs supérieurs comme critère préclinique. Les médicaments en développement pour la SLA ont historiquement utilisé l'extension de la durée de vie de la souris ou la survie des motoneurones inférieurs comme indicateur principal. Or, beaucoup de ces molécules ont échoué chez l'humain. Genç et al. proposent un changement de paradigme : cibler les neurones qui dégénèrent réellement (et précocement) chez les patients, et utiliser des indicateurs cellulaires (mitochondries, RE, dendrites) plutôt que comportementaux ou de survie.
Si NU-9 ne sauve pas les motoneurones inférieurs, à quoi sert-il ?
D'abord, à valider l'hypothèse qu'il est possible de stopper la dégénérescence d'un type de neurone moteur dans la SLA — ce qui n'avait jamais été démontré. Ensuite, parce que les neurones moteurs supérieurs jouent un rôle crucial dans le contrôle moteur volontaire, et qu'ils sont impliqués dans plusieurs autres maladies (sclérose latérale primitive, paraplégie spastique héréditaire, ALS/FTLD). Enfin, dans une stratégie thérapeutique combinée : NU-9 préserverait les UMN tandis que d'autres molécules ciblent les LMN. Des données ultérieures (2022) montrent même qu'NU-9 augmente l'efficacité du riluzole et de l'edaravone sur la régénération axonale.
NU-9 peut-il être efficace dans d'autres maladies neurodégénératives ?
Possiblement. La logique scientifique est solide : la dysfonction mitochondriale et l'agrégation protéique sont des mécanismes partagés par de nombreuses maladies neurodégénératives, dont la maladie d'Alzheimer, la maladie de Parkinson, la dégénérescence frontotemporale (FTLD) et la maladie de Huntington. En mai 2025, une publication dans PNAS a montré qu'AKV9 améliore la mémoire dans un modèle murin de maladie d'Alzheimer (3 souris traitées sur 4 retrouvent des performances de mémoire normales). Akava Therapeutics a également obtenu un financement de 7,3 M$ du National Institute on Aging en 2024 pour étendre les recherches au-delà de la SLA.
🍪 Ce site utilise Google Analytics et le Pixel Facebook pour mesurer son audience. Ces outils ne sont activés qu'avec votre consentement. Politique de confidentialité →