À sa naissance en 2024, KJ Muldoon était atteint d'une maladie génétique ultra-rare qui tue la moitié des bébés dans les premières semaines. En 2025, une équipe américaine a fait quelque chose d'inédit : en six mois, elle a conçu un médicament CRISPR sur mesure pour cet enfant unique au monde. Aujourd'hui, KJ va bien — et la médecine vient de franchir un cap.
Un bébé, un médicament, six mois
À sa naissance en août 2024, KJ Muldoon n'était pas censé vivre longtemps. L'enfant est né avec une maladie génétique qui touche environ une personne sur 1,3 million dans le monde : un déficit en CPS1. Une seule lettre de son ADN, mal orthographiée, empêche son foie d'éliminer l'ammoniac — un déchet produit quand notre corps digère les protéines.
Sans traitement, l'ammoniac s'accumule, le cerveau s'abîme, la moitié des bébés atteints de la forme sévère meurent dans les premières semaines. Les options conventionnelles sont mauvaises : un régime hyper-restrictif permet de survivre quelques mois, une greffe de foie n'est envisageable qu'après plusieurs mois de vie.
À l'Hôpital des enfants de Philadelphie, une équipe a fait quelque chose d'inédit. En six mois, ils ont conçu, fabriqué et administré un médicament sur mesure pour lui seul. Le médicament, c'est CRISPR.
C'est quoi, CRISPR ?
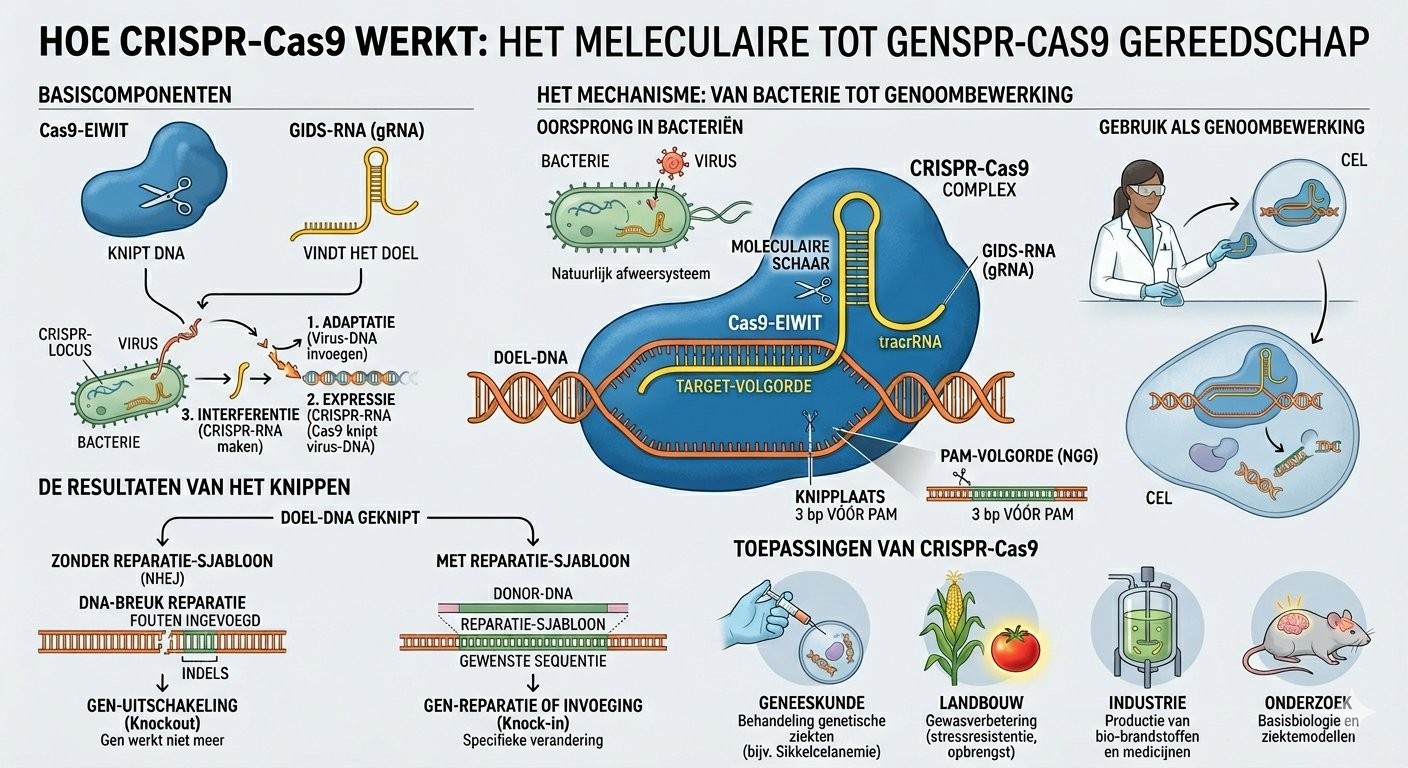
Imagine ton ADN comme une recette de cuisine immense. Trois milliards de lettres qui disent à ton corps comment se construire. Quand une seule lettre est de travers — comme une faute de frappe dans la recette — tout le plat peut être raté.
CRISPR, c'est une paire de ciseaux moléculaires. Mais pas n'importe lesquels : des ciseaux avec un GPS intégré. Le GPS, c'est un petit brin d'ARN (le cousin chimique de l'ADN) qui sait lire la recette et trouver exactement la phrase à corriger. Une fois sur place, une protéine appelée Cas9 vient couper. La cellule se charge de recoller. Et parfois, si on la guide bien, elle peut recoller avec la bonne lettre.
Cette technologie existe depuis 2012. Elle a valu le Prix Nobel de chimie 2020 à Emmanuelle Charpentier et Jennifer Doudna. Ce qui a changé en 2025 avec le cas du bébé KJ, c'est qu'on l'a utilisée pour la première fois pour soigner un seul patient unique au monde.
Pourquoi c'est un virage historique
Jusqu'ici, les médicaments étaient pensés pour des millions de personnes. Développer un traitement prend dix ans et coûte environ un milliard. Pour les maladies ultra-rares — celles qui ne touchent qu'une poignée d'individus dans le monde — aucun laboratoire n'a de raison économique de développer quoi que ce soit.
Le cas KJ prouve qu'on peut aller beaucoup plus vite. Et que le futur de la médecine, peut-être, ce n'est plus un médicament pour tous, mais un médicament pour chacun.
🔍 À retenir
→KJ Muldoon est le premier humain traité avec une thérapie CRISPR conçue spécifiquement pour lui
→Sa maladie (déficit CPS1) touche une personne sur 1,3 million — aucun laboratoire ne développerait de médicament spécifique
→Le traitement a été conçu, fabriqué et administré en 6 mois — contre 10 ans habituellement
→KJ est sorti de l'hôpital en juin 2025 et grandit normalement aujourd'hui
✦ Pour conclure
Le futur de la médecine s'écrit peut-être un patient à la fois.
Le bébé est rentré chez lui le 3 juin 2025, après 307 jours d'hospitalisation. Il mange plus de protéines qu'avant. Il a besoin de moins de médicaments. Il grandit. Ce que le cas KJ raconte, ce n'est pas juste une belle histoire médicale — c'est la preuve que la génétique a franchi un cap. Jusqu'ici, les maladies rares étaient orphelines : trop peu de patients pour intéresser les laboratoires. Avec cette démonstration, la question n'est plus si on peut traiter sur mesure, mais combien de temps et combien d'argent il faudra pour rendre ça accessible à d'autres enfants.
Tu veux le prochain article directement dans ta boîte mail ?
Gratuit · Zéro spam · Toutes les deux semaines.
✦ Résumé intermédiaire
KJ Muldoon, né en 2024 avec un déficit ultra-rare en CPS1 (une enzyme du cycle de l'urée), est devenu en 2025 le premier patient traité par une thérapie CRISPR entièrement personnalisée. L'équipe de Musunuru et Ahrens-Nicklas à Philadelphie a utilisé l'édition de base — une évolution plus précise du CRISPR-Cas9 classique — pour corriger la mutation spécifique de KJ. De la conception à l'infusion : 6 mois. Publié dans le NEJM en mai 2025.
CRISPR-Cas9 : de la défense bactérienne à la clinique
CRISPR n'a pas été inventé. Il a été emprunté. Pendant des millions d'années, les bactéries l'ont utilisé pour se défendre contre les virus. Quand un virus attaque, certaines bactéries conservent un fragment de son ADN dans une zone de leur génome appelée CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats). Si le virus revient, une protéine nommée Cas9, guidée par une copie ARN de ce fragment, le repère et le coupe en deux.
En 2012, Emmanuelle Charpentier et Jennifer Doudna publient dans Science un article qui change tout : elles démontrent qu'on peut reprogrammer ce système en remplaçant l'ARN guide par n'importe quelle séquence. Dès lors, CRISPR-Cas9 n'est plus un mécanisme de défense bactérien : c'est un outil d'édition génomique universel, applicable à toute cellule vivante.
Décembre 2023 : le premier médicament CRISPR approuvé
Treize ans après la publication fondatrice, la FDA américaine et l'agence britannique UKMHRA approuvent Casgevy (exa-cel), développé par Vertex Pharmaceuticals et CRISPR Therapeutics. Ce traitement soigne la drépanocytose et la bêta-thalassémie.
Le principe : on prélève les cellules souches du patient, on les édite hors du corps (ex vivo) pour réactiver un gène inactif depuis l'enfance (celui de l'hémoglobine fœtale), puis on les réinjecte. Résultat chez les patients traités : disparition des crises douloureuses qui caractérisent la drépanocytose.
Mais Casgevy reste un traitement « de masse » : le même protocole pour tous. Le cas KJ inaugure autre chose.
Le bébé KJ : médecine n-of-1
KJ Muldoon est né en août 2024. Quelques jours après sa naissance, son taux d'ammoniac sanguin dépasse 1000 µmol/L — la norme se situe entre 9 et 33. Le diagnostic tombe : déficit sévère en carbamoyl phosphate synthétase 1 (CPS1), une enzyme hépatique du cycle de l'urée. Sans elle, l'ammoniac ne peut pas être transformé en urée et évacué. Il s'accumule. Il attaque le cerveau.
Kiran Musunuru (Université de Pennsylvanie) et Rebecca Ahrens-Nicklas (Children's Hospital of Philadelphia) décident de tenter autre chose. Ils identifient les deux mutations précises du gène CPS1 de KJ. Puis ils conçoivent une thérapie spécifique à ces mutations. Le tout en six mois, de la conception à l'infusion.
La clé : ils n'ont pas utilisé un CRISPR-Cas9 classique. Ils ont utilisé une évolution plus récente et plus précise : l'édition de base (base editing).
L'édition de base : CRISPR en plus fin
Le CRISPR classique cause des cassures double-brin de l'ADN. C'est efficace, mais risqué : la cellule peut recoller mal, et parfois créer des réarrangements chromosomiques importants.
L'édition de base ne coupe pas. Elle convertit chimiquement une lettre de l'ADN en une autre — par exemple un A en G — directement sur place, sans cassure. Pour le cas KJ, l'équipe a conçu une édition qui corrige une des deux mutations responsables de son déficit, juste assez pour restaurer un niveau minimal d'enzyme fonctionnelle.
Le traitement a été livré au foie via des nanoparticules lipidiques (les mêmes technologies que les vaccins à ARN COVID). Trois doses, entre février et avril 2025.
Aujourd'hui : ce qu'on sait, ce qu'on ne sait pas
Ce qu'on sait : KJ est sorti de l'hôpital 307 jours après son admission. Il peut manger plus de protéines qu'avant. Il a besoin de moins de médicaments réducteurs d'ammoniac. Il grandit normalement.
Ce qu'on ne sait pas : combien de temps l'effet va durer, si l'édition va se maintenir à mesure que ses cellules hépatiques se renouvellent, et si des effets hors cible se révéleront dans les années à venir.
🔍 Points clés
→CRISPR-Cas9 = système immunitaire bactérien reprogrammé comme outil d'édition universelle (Jinek et al., Science 2012)
→Casgevy (décembre 2023) : premier médicament CRISPR approuvé, pour drépanocytose et β-thalassémie — approche ex vivo, même protocole pour tous
→Cas KJ (février 2025) : première thérapie CRISPR « N-of-1 » — base editing in vivo via nanoparticules lipidiques, ciblant une mutation unique
→Délai historique compressé : 6 mois de la conception à l'infusion, vs ~10 ans pour un médicament classique
✦ Pour conclure
Prototype d'un modèle, pas exception isolée.
D'après l'Innovative Genomics Institute, environ 250 essais cliniques mondiaux utilisent aujourd'hui des outils d'édition génomique, dont plus de 150 actifs. La plupart testent encore des protocoles uniformes pour groupes de patients. Mais deux programmes américains lancés en 2025 (THRIVE et GIVE de l'ARPA-H) visent explicitement à rendre réplicable l'approche « N-of-1 » : construire les infrastructures réglementaires et industrielles permettant de compresser 10 ans de développement en 6 mois pour n'importe quel enfant atteint d'une mutation ultra-rare. Si ça marche, c'est un changement de paradigme. Si ça ne marche pas, KJ restera une exception magnifique mais isolée. La suite se joue ces trois prochaines années.
Musunuru, Ahrens-Nicklas et al. (NEJM, 2025) rapportent le premier cas clinique d'une thérapie d'édition génomique personnalisée N-of-1 chez un nourrisson atteint de déficit sévère en carbamoyl phosphate synthétase 1 (CPS1). L'équipe a conçu un adénine-base éditeur (ABE) ciblant les variants tronquants spécifiques du patient, délivré via nanoparticules lipidiques en trois doses systémiques (février–avril 2025). Le pipeline — de l'identification mutationnelle à l'infusion — a été complété en ~6 mois. Le patient tolère mieux les apports protéiques, a vu ses besoins en scavengers ammoniacaux diminuer, et a été externalisé après 307 jours d'hospitalisation.
Architecture moléculaire du traitement KJ
Le traitement administré à KJ n'est pas un CRISPR-Cas9 conventionnel. C'est un adénine-base éditeur (ABE), une architecture dérivée de CRISPR mais fondamentalement différente dans son mécanisme d'action.
Un ABE est constitué de trois modules fusionnés :
Une Cas9 « nickase » (nCas9) dont une des deux activités nucléasiques (HNH) est inactivée par mutation ponctuelle. Elle n'entaille qu'un seul brin de l'ADN au lieu des deux — évitant la cassure double-brin.
Une adénine désaminase évoluée (ABE8e), qui convertit chimiquement une adénine (A) en inosine (I, lue comme une guanine G par la machinerie cellulaire).
Un ARN guide de 20 nucléotides complémentaire à la séquence cible, adjacent à un PAM (motif NGG pour la Cas9 de Streptococcus pyogenes).
Concrètement : la cible est localisée par appariement ARN-ADN, un seul brin est entaillé, et la désaminase modifie chimiquement une A spécifique dans une fenêtre d'édition d'environ 4-8 nucléotides. Après réplication, le brin opposé est recopié à partir du brin modifié, fixant la mutation A→G de façon permanente.
Dans le cas KJ, l'équipe Musunuru a ciblé les mutations tronquantes du gène CPS1 et conçu un ARN guide permettant une conversion A→G restaurant la lecture correcte de l'exon concerné. La publication NEJM (DOI : 10.1056/NEJMoa2504747) détaille le design et les tests précliniques rapides en souris humanisées avant l'administration chez le patient.
Pourquoi le base editing change la donne
La différence cruciale entre CRISPR-Cas9 classique et base editing réside dans le profil de sécurité. Les grandes délétions et réarrangements chromosomiques se produisent à une fréquence environ 20 fois plus faible avec les base editors qu'avec les nucléases Cas9 classiques.
C'est essentiel, car les risques majeurs du CRISPR-Cas9 conventionnel ne sont pas seulement les petites insertions-délétions (indels) sur les sites hors cible, mais aussi des variations structurelles plus larges : délétions de plusieurs mégabases, translocations chromosomiques, et réarrangements complexes au site même d'édition.
Pour un nourrisson de 7 mois, l'équation risque/bénéfice favorisait clairement l'approche la plus conservatrice techniquement, même si elle ciblait une mutation « mineure » du point de vue de la correction (restauration partielle d'activité plutôt que correction complète).
Délivrance : la contrainte limitante
La thérapie de KJ a été délivrée par nanoparticules lipidiques (LNP) — la même plateforme que les vaccins à ARNm contre le SARS-CoV-2. Les LNP encapsulent l'ARNm codant l'éditeur de base et l'ARN guide, et s'accumulent préférentiellement dans le foie après administration IV, ce qui est parfait quand la cible (ici, les hépatocytes qui expriment CPS1) est hépatique.
Mais c'est aussi une limite structurelle du modèle. Les LNP hépatotropes fonctionnent bien pour le foie — d'où la multiplication des essais sur les maladies hépatiques (hATTR avec NTLA-2001 d'Intellia, AATD-1, GSD1a avec Beam Therapeutics). Pour d'autres tissus (cerveau, muscle, rétine), il faut d'autres vecteurs : AAV principalement, avec des défis d'immunogénicité et de capacité de cargo (les AAV plafonnent autour de 4,7 kb, ce qui est serré pour un éditeur de base complet).
L'état clinique mondial en 2026
D'après l'Innovative Genomics Institute (mise à jour 2026), environ 250 essais cliniques mondiaux utilisent l'édition génomique (CRISPR-Cas, base editing, prime editing, ZFN, TALEN), dont plus de 150 actifs. Les aires thérapeutiques principales :
Oncologie : essais CAR-T enrichis par édition (KO de PD-1, TRAC, etc.) — majoritairement phase 1/2.
Le cas KJ n'est donc pas un outlier isolé. Il s'inscrit dans un déplacement du paradigme clinique : de la thérapie « de plateforme » (Casgevy) vers la thérapie « bespoke » (N-of-1). L'ARPA-H américaine a lancé en 2025 deux programmes (THRIVE et GIVE) destinés à créer les infrastructures réglementaires et manufacturières permettant de répliquer l'approche KJ pour d'autres patients.
🔬 Limitations & perspectives
→Durabilité de l'édition : les hépatocytes se renouvellent. Si l'édition n'a touché qu'une fraction des cellules souches hépatiques, l'effet thérapeutique pourrait s'éroder. Suivi à long terme crucial.
→Mosaïcisme : tous les hépatocytes ne sont pas édités. Le ratio d'édition n'a pas été publié en détail — une correction partielle a suffi pour restaurer une fonction métabolique compatible avec la vie.
→Effets hors cible sgRNA-indépendants : les ABE8e présentent un profil d'édition ARN potentiellement problématique. Monitoring transcriptomique nécessaire dans le suivi pédiatrique.
→Scalabilité économique : traitement à plusieurs millions USD, pris en charge par CHOP et ses partenaires philanthropiques. Aucun modèle de remboursement n'existe pour les N-of-1 — c'est le goulot d'étranglement réel.
→Cadre réglementaire : la FDA a autorisé le traitement sous protocole de recherche exceptionnel. Création de voies pérennes = enjeu 2026-2028.
✦ Pour conclure
Le vrai saut n'est pas technologique — il est organisationnel.
CRISPR n'est pas « la médecine du futur ». C'est la médecine d'aujourd'hui — mais pas au sens où beaucoup le croient. L'imaginaire public reste fixé sur les « bébés génétiquement modifiés » (épisode He Jiankui, 2018) ou sur les fantasmes d'amélioration génétique. La réalité clinique est plus prosaïque et plus radicale : CRISPR a déjà sauvé des vies chez des patients souffrant de drépanocytose, et il vient de prouver qu'il peut être mobilisé en six mois pour sauver un bébé unique au monde. L'équipe a démontré qu'avec une coordination étroite entre laboratoire, fabricants de LNP, CROs, et autorités réglementaires, on peut compresser un processus qui prend habituellement une décennie en 180 jours. Ce chaînage, s'il devient réplicable, transformera le traitement des maladies ultra-rares. La question n'est plus technique. Elle est économique, réglementaire, et organisationnelle.
Source primaire
Musunuru K, Ahrens-Nicklas R, et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. New England Journal of Medicine. 2025. doi:10.1056/NEJMoa2504747. Projet conduit par Children's Hospital of Philadelphia et Penn Medicine avec partenaires philanthropiques.
Références clés
[1]Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/science.1225829 — Papier fondateur du CRISPR-Cas9 programmable.
Combien coûte un traitement CRISPR personnalisé comme celui du bébé KJ ?
Le coût exact n'a pas été rendu public, mais est estimé à plusieurs millions de dollars. Le traitement du bébé KJ a été pris en charge par le Children's Hospital of Philadelphia (CHOP), Penn Medicine et leurs partenaires philanthropiques. Aucun modèle de remboursement standardisé n'existe actuellement pour les thérapies dites « N-of-1 » — c'est précisément le goulot d'étranglement qui empêche sa généralisation à d'autres patients.
CRISPR peut-il modifier l'ADN de nos enfants (modifications héréditaires) ?
Techniquement, oui — mais c'est strictement interdit en clinique dans la quasi-totalité des pays. Le traitement du bébé KJ modifie uniquement les cellules du foie (cellules somatiques) : ces modifications ne seront pas transmises à sa descendance. L'édition germinale (embryons, ovules, spermatozoïdes) reste proscrite depuis l'épisode He Jiankui en 2018, qui avait suscité une condamnation unanime de la communauté scientifique internationale.
Quelles sont les maladies actuellement soignées avec CRISPR en 2026 ?
Un seul médicament CRISPR est officiellement approuvé : Casgevy (exa-cel), pour la drépanocytose et la bêta-thalassémie, approuvé fin 2023 aux États-Unis et au Royaume-Uni. Environ 250 essais cliniques sont en cours dans le monde (dont plus de 150 actifs), couvrant l'amyloïdose hATTR, l'angio-œdème héréditaire, les maladies métaboliques rares et certains cancers. La plupart sont encore en phase 1 ou 2. Le cas KJ est unique : c'est le premier traitement conçu spécifiquement pour un seul patient.
CRISPR peut-il créer des « bébés sur mesure » comme dans les films ?
Non, et c'est un mythe important à déconstruire. Les applications cliniques actuelles de CRISPR ciblent des cellules adultes déjà malades pour corriger des mutations pathologiques précises. La sélection de caractéristiques comme la couleur des yeux, l'intelligence ou la taille reste totalement hors de portée scientifique (la plupart de ces traits sont polygéniques, contrôlés par des centaines de gènes) et strictement illégale sur le plan éthique. Le vrai enjeu de CRISPR aujourd'hui, ce ne sont pas les bébés sur mesure, mais le traitement de maladies génétiques graves.
Pourquoi la thérapie du bébé KJ ne peut pas être facilement dupliquée ?
Chaque mutation génétique est unique. L'ARN guide qui a corrigé le gène CPS1 de KJ ne fonctionnerait pas pour un autre enfant avec une mutation différente, même dans le même gène. Il faut donc redessiner l'ARN guide, retester la thérapie en laboratoire, produire une formulation adaptée et obtenir une autorisation réglementaire — et répéter ce processus pour chaque patient. Les programmes américains THRIVE et GIVE (ARPA-H, 2025) visent justement à industrialiser ce pipeline pour qu'il devienne reproductible en quelques mois.
Qu'est-ce que l'édition de base (base editing), et pourquoi est-ce différent du CRISPR classique ?
Le CRISPR-Cas9 classique coupe l'ADN des deux côtés, puis s'appuie sur la cellule pour réparer la cassure — ce qui peut provoquer des réarrangements chromosomiques. L'édition de base, développée par David Liu en 2016, ne coupe pas : elle convertit chimiquement une lettre d'ADN en une autre (par exemple un A en G) directement sur place. C'est environ 20 fois plus sûr en termes de grandes délétions et translocations. C'est cette version qui a été utilisée pour traiter le bébé KJ.